
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuaternary β2,2-amino acid derivatives by asymmetric addition of isoxazolidin-5-ones to para-quinone methides†
Andreas
Eitzinger
,
Michael
Winter
,
Johannes
Schörgenhumer
 and
Mario
Waser
*
and
Mario
Waser
*
Johannes Kepler University Linz, Institute of Organic Chemistry, Altenbergerstraße 69, 4040 Linz, Austria. E-mail: mario.waser@jku
First published on 6th December 2019
Abstract
The highly enantioselective (>99.5% ee) synthesis of a new class of densely functionalized β2,2-amino acid derivatives by reacting isoxazolidin-5-ones with para-quinone methides in the presence of chiral ammonium salt phase-transfer catalysts was developed. The reaction proceeds with exceptionally low catalyst loadings down to 20 ppm on gram scale and the utilization of the primary addition products towards further manipulations was demonstrated for selected examples.
Chiral β-amino acids (AA) are very interesting structural motifs which have attracted significant attention over the last decades.1–4 Their high value is because β-AA-containing compounds show unique biological properties combined with increased metabolic stability and, in addition, it turned out that their incorporation into peptides results in well-defined and more rigid secondary structures, compared to α-AA-based ones.1,2 It is thus not surprising that the development of novel synthesis methods to access those valuable targets has become a very important topic.3,4 While syntheses of β-AA containing only one substituent in the α- and/or β-position (β2, β3 or β2,3-AA) have been well-established,3,4 the asymmetric syntheses of β-AA containing an all-carbon α-quaternary stereocenter but no further substituents in the β-position (β2,2-AA) remain a synthetic challenge.5–8
An elegant and straightforward strategy to access masked β2,2-AA in an asymmetric catalytic manner relies on the use of easily accessible isoxazolidin-5-ones 1 as pronucleophiles (Scheme 1A).7,8 These compounds can be directly accessed from Meldrum acid derivatives via an elegant route developed by Briere and co-workers.9 The same group also pioneered the use of compounds 1 for asymmetric transformations to access α-sulfanylated, α-aminated, and α-alkylated derivatives8 under asymmetric phase-transfer catalysis (PTC).10 In addition, they demonstrated the utilization of the hereby obtained products towards chiral β2,2-AA derivatives (i.e. by reductive cleavage of the N–O bond), giving access to chiral β2,2-AA that are not accessible by other common strategies. Shortly after these initial reports the groups of Shibasaki and Cossy independently reported transition metal-catalysed asymmetric α-allylation reactions of compounds 1,7a,b as well as organocatalytic Michael and Mannich reactions.7c In addition Noda and Shibasaki recently also demonstrated that isoxazolidinones 1 can undergo intramolecular electrophilic aromatic aminations (by N–O bond cleavage), providing another powerful application for these unique compounds.11 Our group has a long-standing interest in asymmetric PTC,12 and we recently reported the enantioselective addition of 1 to MBH carbonates in the presence of chiral PTCs.7d However, apart from those few very recent reports describing the utilization of pronucleophiles 1 to access (masked) all-carbon quaternary β2,2-AA,7,8 no further asymmetric approaches relying on the use of compounds 1 have been reported so far (to the best of our knowledge). We thus wondered if we would be able to develop a broadly applicable and highly stereoselective method to access a new family of densely functionalized (masked) β2,2-AA. We were especially interested in the synthesis of novel β′,β′-diarylated-β2,2-amino acids as it was recently shown that β,β-diarylated-α-AA possess very promising biological properties,13 and we thus reasoned that the so far unknown homologous β-AA would be worthwhile targets.
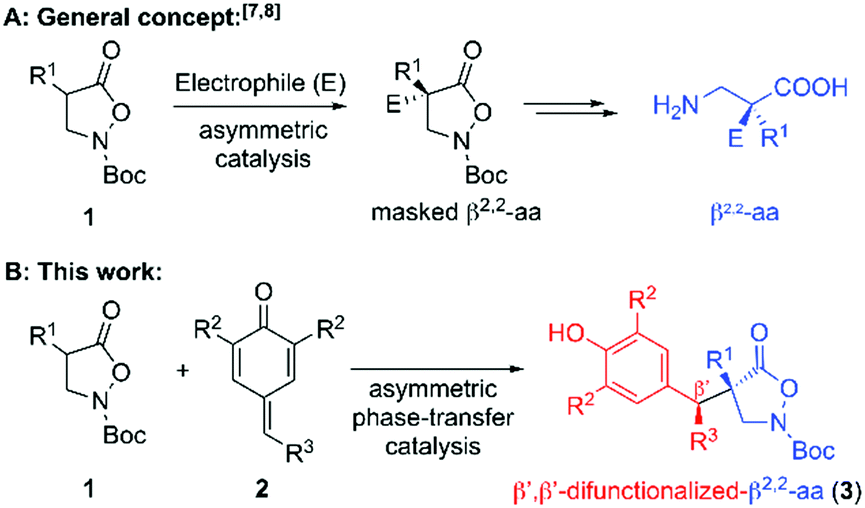 | ||
| Scheme 1 Asymmetric β2,2-AA syntheses starting from isoxazolidin-5-ones 1. | ||
One class of acceptor molecules that turned out to be highly versatile are p-quinone methides (p-QMs; 2).14 These easily accessible and reasonably electrophilic15 compounds have recently emerged as outstanding acceptors to access high levels of structural complexity upon reaction with different (pro)-nucleophiles16,17 and we reasoned that the development of a stereoselective catalytic protocol for the addition of masked β-AA 1 to p-QMs 2 would result in a unique and powerful approach to access a new class of highly functionalized dissymmetric β′,β′-difunctionalized-β2,2-AA 3 (Scheme 1B).
We started by carrying out the reaction between the α-benzyl isoxazolidin-5-one 1a and the parent p-quinone methide 2a in the presence of a variety of different achiral and chiral phase-transfer catalysts (Table 1 gives the most significant results).18 First racemic experiments using K2CO3 and Cs2CO3 showed that Cs2CO3 alone allows for product 3a formation (entry 2), while K2CO3 requires the addition of a quaternary ammonium salt to promote the target reaction (entries 1 and 3). To suppress the uncatalyzed background reaction as good as possible, we thus carried out the further screening using K2CO3 first (entries 4–8). In analogy to our recent observations when reacting compounds 1 with MBH carbonates,7d and Briere's results when using 1 under asymmetric phase-transfer catalysis,8 the only catalysts that allowed for high selectivities were Maruoka's commercially available binaphthyl-based spiro ammonium salts A.18,19 Those gave promising enantioselectivities already under the unoptimized conditions (entries 4 and 5), with the 3,4,5-trifluorophenyl-based A1 being slightly better suited then A2. Noteworthy, the reaction also gave high enantioselectivities when using a catalytic amount of base, albeit processing significantly slower (entry 6). In order to improve diastereo- and enantioselectivity, we screened different solvents and bases and found that dioxane in combination with a catalytic amount of either K2CO3 or Cs2CO3 allows for high enantio- and moderate diastereoselectivities (entries 7 and 9). The high selectivity obtained with Cs2CO3 was especially encouraging, as it demonstrates a very high catalytic activity when considering the fact that the reaction proceeds well in the absence of ammonium salts as well (compare with entry 2). Unfortunately, reactions with catalytic amounts of base turned out to be rather slow (requiring >72 h to complete) and in general reactions using Cs2CO3 were found to be more robust and better reproducible (compared to other bases).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Solv. | Base (eq.) | Yieldb [%] | drc | eed |
| a All reactions were run for 24 h at room temperature unless otherwise stated using 0.1 mmol 1a, 0.15 mmol 2a and 5 mol% of the catalyst (0.1 M with respect to 1a). b Isolated yields. c Determined by 1H NMR of the crude product. d Determined by HPLC using a chiral stationary phase. e Triethylbenzylammonium chloride. f Reactions had to be run for more than 72 h to ensure full conversion. g Run at −20 °C. | ||||||
| 1 | — | THF | K2CO3 (1.1) | <5 | — | — |
| 2 | — | THF | Cs2CO3 (1.1) | 89 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.7 :1.7 |
— |
| 3 | TEBACe | THF | K2CO3 (1.1) | 94 | 1:1.6 |
— |
| 4 | A1 | THF | K2CO3 (1.1) | 90 | 2.7:1 |
93 |
| 5 | A2 | THF | K2CO3 (1.1) | 91 | 1.8:1 |
90 |
| 6f | A1 | THF | K2CO3 (0.2) | 28 | 3.2:1 |
95 |
| 7f | A1 | Dioxane | K2CO3 (0.2) | 92 | 5.4:1 |
99 |
| 8f | A1 | Et2O | K2CO3 (0.2) | 91 | 4.8:1 |
95 |
| 9f | A1 | Dioxane | Cs2CO3 (0.1) | 94 | 4.5:1 |
98 |
| 10g | A1 | Et2O | Cs2CO3 (1.1) | 97 | 10:1 |
>99.5 |
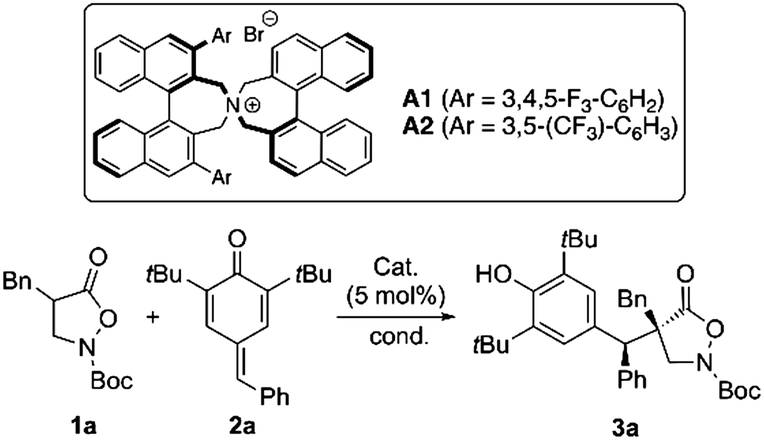
In order to improve ee and especially dr further, we tested lower temperatures as well. However, as the melting point of dioxane made lowering the reaction temperature not possible, we changed for Et2O in those experiments. Gratifyingly, we finally found that the use of a stoichiometric amount of Cs2CO3 at −20 °C (24 h) gives product 3a with excellent enantioselectivity (>99.5% ee), high diastereoselectivity (10:1), and in literally quantitative yield (97%) (entry 10, please note that the uncatalyzed background reaction becomes significantly slower at −20 °C compared to room temperature conditions).
With these operationally simple conditions at hand, we next investigated the influence of the catalyst loading (Scheme 218). As the Maruoka catalyst A1 is only rather sparingly soluble in Et2O we reasoned that the undissolved catalyst may only serve as a reservoir for the catalytically relevant dissolved ammonium salt. In addition, given the high selectivities observed when using this catalyst under conditions where the uncatalyzed racemic background is relatively fast as well, we were hoping that we could reduce the catalyst loading significantly. Remarkably, we were able to carry out the reaction with exceptionally low catalyst loadings of down to 0.002 mol% (20 ppm), without significantly affecting the selectivity. We only observed that the conversion became slightly slower, requiring 48 h of reaction time to give 90% isolated yield for this 20 ppm experiment, while “higher” catalyst loadings resulted in full conversions after 12–24 h.18 Autocatalysis could be ruled out by control experiments and thus this high selectivity is really a consequence of a very remarkable catalyst control herein. To the best of our knowledge, this is one of the lowest asymmetric phase-transfer catalyst loadings reported so far, resulting in an efficient process to access the target 3a with very high ee.20
 | ||
| Scheme 2 Low catalyst loading gram scale synthesis of almost enantiopure 3a. | ||
To demonstrate the generality of this reaction we next investigated the application scope by using a broad variety of differently substituted pronucleophiles 1 and acceptors 2 (Table 2). We first screened a variety of differently substituted nucleophiles 1 (entries 1–14). All of them performed similarly well, giving access to the products 3a–3n (differing in their R1 substituents) in almost quantitative yields and with excellent enantioselectivities and high diastereoselectivities. To elucidate the relative and the absolute configuration of the newly formed products 3 we performed X-ray analysis of single crystals of the major stereoisomer of the bromo-derivative 3j. This allowed for an unambiguous elucidation of the configuration of this derivative and the configuration of the other targets was assigned in analogy (we also carried out X-ray analysis of single crystals of racemic 3b confirming the relative configuration of this derivative as well).21 Next, we varied the quinone methide acceptors 2 (entries 15–25). Besides the bis-t-butyl-containing QMs also differently R2-substituted derivatives, as shown in entries 15 and 16, can be successfully employed with high selectivities and high yields. Alternative aromatic R3 groups were well accepted too, as outlined in entries 17–23. Here we also carried out one experiment with N-Cbz-protected 1a (entry 20), which resulted in a similarly high enantioselectivity (compared to the parent N-Boc protected derivative), but gave the product in lower yield, mainly because of the formation of notable amounts of unidentified side-products. We were also pleased to see that vinylogous QMs were equally well tolerated too (entry 24). Finally, one novel acceptor that we were especially interested in is the CF3-containing QM 2x, which we herein report for the first time22 as a versatile building block for asymmetric catalysis. The use of this unique QM allows for the synthesis of the CF3-containing masked β-aa 3x in very high selectivity as well (entry 25), giving access to an interesting new class of CF3-containing β-amino acid derivatives.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | 3 [%] | drc | eed,e |
| a For ease of comparison all reactions were run for 24 h at −20 °C in Et2O (0.1 M with respect to 1) using 0.1 mmol 1, 0.15 mmol 2, 0.11 mmol Cs2CO3, and 5 mol% of A1 (selected examples were repeated using 1 mol% A1 without affecting the outcome). b Isolated yields. c Determined by NMR of the crude product. d Determined by HPLC using a chiral stationary phase. e The relative and absolute configuration of 3j was proven by X-ray single crystal diffraction analysis23 and all other compounds were assigned in analogy. f Using N-Cbz-protected 1a. | ||||||
| 1 | Bn | t-Bu | Ph- | 97 (3a) | 10:1 |
>99.5 |
| 2 | 4-Ph-C6H4-CH2- | t-Bu | Ph- | 99 (3b) | 10:1 |
>99.5 |
| 3 | 2-Naphthyl-CH2- | t-Bu | Ph- | 97 (3c) | 10:1 |
>99.5 |
| 4 | 2-Furanyl-CH2- | t-Bu | Ph- | 98 (3d) | 11:1 |
>99.5 |
| 5 | 4-Me-C6H4-CH2 | t-Bu | Ph- | 97 (3e) | 10:1 |
>99.5 |
| 6 | 2-Me-C6H4-CH2 | t-Bu | Ph- | 96 (3f) | 3:1 |
>99.5 |
| 7 | 4-t-Bu-C6H4-CH2 | t-Bu | Ph- | 94 (3g) | 14:1 |
>99.5 |
| 8 | 4-MeO-C6H4-CH2 | t-Bu | Ph- | 99 (3h) | 9:1 |
>99.5 |
| 9 | 4-Cl-C6H4-CH2 | t-Bu | Ph- | 97 (3i) | 8:1 |
99.3 |
| 10e | 4-Br-C6H4-CH2 | t-Bu | Ph- | 95 (3j) | 7:1 |
99.4 |
| 11 | 4-F-C6H4-CH2 | t-Bu | Ph- | 99 (3k) | 7:1 |
99.1 |
| 12 | (E)-Ph–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2 CH–CH2 |
t-Bu | Ph- | 95 (3l) | 9:1 |
99.4 |
| 13 | Cyclopropyl-CH2 | t-Bu | Ph- | 90 (3m) | 9:1 |
98.0 |
| 14 | Cyclohexyl-CH2 | t-Bu | Ph- | 91 (3n) | 7:1 |
>99.5 |
| 15 | Bn | i-Pr | Ph- | 90 (3o) | 5:1 |
98.9 |
| 16 | Bn | t-Bu, Me | Ph- | 99 (3p) | 10:1 |
99.5 |
| 17 | Bn | t-Bu | 4-MeO-C6H4- | 95 (3q) | 6:1 |
99.1 |
| 18 | Bn | t-Bu | 2-MeO-C6H4- | 99 (3r) | 6:1 |
>99.5 |
| 19 | Bn | t-Bu | 4-Cl-C6H4- | 99 (3s) | 20:1 |
>99.5 |
| 20f | Bn | t-Bu | 4-Cl-C6H4- | 35 (3s-Cbz) | 14:1 |
99.0 |
| 21 | Bn | t-Bu | 4-CF3-C6H4- | 99 (3t) | 6:1 |
>99.5 |
| 22 | Bn | t-Bu | 2-Naphthyl- | 99 (3u) | 11:1 |
>99.5 |
| 23 | Bn | t-Bu | 2-Pyridyl- | 97 (3v) | 4:1 |
96.5 |
| 24 | Bn | t-Bu | (E)-Ph-CHCH- |
70 (3w) | 5:1 |
98.5 |
| 25 | Bn | t-Bu | CF3- | 97 (3x) | 10:1 |
>99.5 |

To demonstrate the versatility of products 3 for further manipulations we carried out the test reactions shown in Scheme 3. The N–O-bond could easily be cleaved under Pd-catalyzed hydrogenation conditions (either using H2 or HCO2NH4)8,9 to get access to the N-protected β2,2-amino acid 4a straightforwardly (Scheme 3A). This compound could then be deprotected and debutylated using AlCl3 (giving 8a). In addition, as demonstrated by Noda and Shibasaki recently,7a isoxazolidinones can be directly employed for KAHA-type ligations.23 We were glad to see that this strategy can also be applied to utilize 3a to access the dipeptide 7a in high isolated yield upon treatment of the in situ formed deprotected isoxazolidinone 5a with β-ketoacid 6 (Scheme 3B).
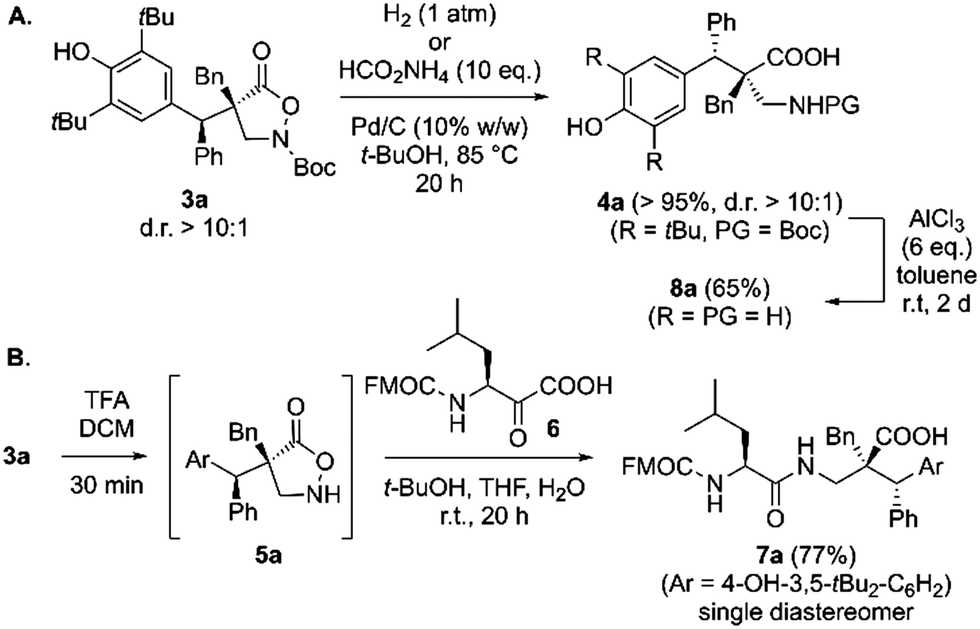 | ||
| Scheme 3 Further transformations of 3a. | ||
In conclusion, we have developed the highly enantioselective (>99.5% ee) synthesis of a new class of densely functionalized β2,2-AA derivatives 3 by reacting isoxazolidin-5-ones 1 with para-quinone methides 2 in the presence of commercially available Maruoka PTCs. The reaction tolerates a broad variety of differently substituted starting materials and proceeds with exceptionally low catalyst loadings down to 0.002 mol% (20 ppm) on gram scale. Furthermore, we demonstrated the utilization of the primary addition products towards further manipulations, like KAHA-type ligations, hydrogenation reactions, and aryl-debutylations.
Financial support by the Austrian Science Funds (FWF), Project No. P30237, is gratefully acknowledged. The used NMR spectrometers were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, “RERI-uasb”).
Conflicts of interest
There are no conflicts to declare.Notes and references
- Selected reviews: (a) R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS PubMed; (b) G. Lelais and D. Seebach, Pept. Sci., 2004, 76, 206–243 CrossRef CAS PubMed; (c) M.-I. Aguilar, A. W. Purcell, R. Devi, R. Lew, J. Rossjohn, A. I. Smith and P. Perlmutter, Org. Biomol. Chem., 2007, 5, 2884–2890 RSC; (d) D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366–1375 CrossRef CAS PubMed; (e) Y.-D. Wu, W. Han, D.-P. Wang, Y. Gao and Y.-L. Zhao, Acc. Chem. Res., 2008, 41, 1418–1427 CrossRef CAS PubMed.
- D. Seebach, S. Abele, T. Sifferlen, M. Hänggi, S. Gruner and P. Seiler, Helv. Chim. Acta, 1998, 81, 2218–2243 CrossRef CAS.
- Enantioselective Synthesis of β-Amino Acids, ed. E. Juaristi and V. A. Soloshonok, 2nd edn, John Wiley & Sons, 2005 Search PubMed.
- Reviews on β-AA syntheses: (a) S. Abele and D. Seebach, Eur. J. Org. Chem., 2000, 1–15 CrossRef CAS; (b) J.-A. Ma, Angew. Chem., Int. Ed., 2003, 42, 4290–4299 CrossRef CAS PubMed; (c) D. Seebach, A. K. Beck, S. Capone, G. Deniau, U. Groselj and E. Zass, Synthesis, 2009, 1–32 CrossRef CAS; (d) B. Weiner, W. Szymanski, D. B. Janssen, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2010, 39, 1656–1691 RSC.
- For selected pioneering reports relying on conjugate additions to β,β-disubstituted nitroalkenes: (a) H.-H. Lu, F.-G. Zhang, X.-G. Meng, S.-W. Duan and W.-J. Xiao, Org. Lett., 2009, 11, 3946–3949 CrossRef CAS PubMed; (b) R. Kastl and H. Wennemers, Angew. Chem., Int. Ed., 2013, 52, 7228–7232 CrossRef CAS PubMed; (c) K. Mori, M. Wakazawa and T. Akiyama, Chem. Sci., 2014, 5, 1799–1803 RSC.
- For a pioneering report relying on the use of α-cyanoacetates: T.-Y. Liu, R. Li, Q. Chai, J. Long, B.-J. Li, Y. Wu, L.-S. Ding and Y.-C. Chen, Chem. – Eur. J., 2007, 13, 319–327 CrossRef CAS PubMed.
- (a) J.-S. Yu, H. Noda and M. Shibasaki, Angew. Chem., Int. Ed., 2018, 57, 818–822 CrossRef CAS PubMed; (b) M. N. de Oliveira, S. Arseniyadis and J. Cossy, Chem. – Eur. J., 2018, 24, 4810–4814 CrossRef PubMed; (c) J.-S. Yu, H. Noda and M. Shibasaki, Chem. – Eur. J., 2018, 24, 15796–15800 CrossRef CAS PubMed; (d) V. Capaccio, K. Zielke, A. Eitzinger, A. Massa, L. Palombi, K. Faust and M. Waser, Org. Chem. Front., 2018, 5, 3336–3340 RSC; (e) F. Amemiya, H. Noda and M. Shibasaki, Chem. Pharm. Bull., 2019, 67, 1046–1049 CrossRef PubMed.
- (a) T. Cadart, C. Berthonneau, V. Levacher, S. Perrio and J.-F. Briere, Chem. – Eur. J., 2016, 22, 15261–15264 CrossRef CAS PubMed; (b) T. Cadart, V. Levacher, S. Perrio and J.-F. Briere, Adv. Synth. Catal., 2018, 360, 1499–1509 CrossRef CAS.
- (a) T. Tite, M. Sabbah, V. Levacher and J.-F. Briere, Chem. Commun., 2013, 49, 11569–11571 RSC; (b) N. Lespes, E. Pair, C. Maganga, M. Bretier, V. Tognetti, L. Joubert, V. Levacher, M. Hubert-Roux, C. Afonso, C. Loutelier-Bourhis and J.-F. Briere, Chem. – Eur. J., 2018, 24, 4086–4093 CrossRef CAS PubMed.
- Recent reviews on asymmetric PTCs: (a) J.-F. Briere, S. Oudeyer, V. Dalla and V. Levacher, Chem. Soc. Rev., 2012, 41, 1696–1707 RSC; (b) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS PubMed; (c) J. Tan and N. Yasuda, Org. Process Res. Dev., 2016, 20, 129–139 CrossRef; (d) L. Zong and C.-H. Tan, Acc. Chem. Res., 2017, 50, 842–856 CrossRef CAS PubMed; (e) J. Schörgenhumer, M. Tiffner and M. Waser, Beilstein J. Org. Chem., 2017, 13, 1753–1769 CrossRef PubMed; (f) H. Wang, Catalysts, 2019, 9, 244 CrossRef.
- (a) J.-S. Yu, M. Espinosa, H. Noda and M. Shibasaki, J. Am. Chem. Soc., 2019, 141, 10530–10537 CrossRef CAS PubMed; (b) M. Espinosa, H. Noda and M. Shibasaki, Org. Lett., 2019, 21, 9296–9299 CrossRef CAS PubMed.
- For a recent example see: J. Novacek, J. A. Izzo, M. J. Vetticatt and M. Waser, Chem. – Eur. J., 2016, 22, 17339–17344 CrossRef CAS PubMed.
- For two examples: (a) M. Nilsson, M. Hämäläinnen, M. Ivarsson, J. Gottfries, Y. Xue, S. Hansson, R. Isaksson and T. Fex, J. Med. Chem., 2009, 52, 2708–2715 CrossRef CAS PubMed; (b) D. E. Patterson, J. D. Powers, M. LeBlanc, T. Sharkey, E. Boehler, E. Irdam and M. H. Osterhout, Org. Process Res. Dev., 2009, 13, 900–906 CrossRef CAS.
- (a) W.-J. Bai, J.-G. David, Z.-G. Feng, M. G. Weaver, K.-L. Wu and T. R. R. Pettus, Acc. Chem. Res., 2014, 47, 3655–3664 CrossRef CAS PubMed; (b) N. J. Willis and C. D. Bray, Chem. – Eur. J., 2012, 18, 9160–9173 CrossRef CAS PubMed; (c) M. S. Singh, A. Nagaraju, N. Anand and S. Chowdhury, RSC Adv., 2014, 4, 55924–55959 RSC; (d) L. Caruana, M. Fochi and L. Bernardi, Molecules, 2015, 20, 11733–11764 CrossRef CAS PubMed; (e) D. V. Osipov, V. A. Osyanin and Y. N. Klimochkin, Russ. Chem. Rev., 2017, 86, 625–687 CrossRef CAS.
- (a) R. Lucius, R. Loos and H. Mayr, Angew. Chem., Int. Ed., 2002, 41, 91–95 CrossRef CAS; (b) D. Richter, N. Hampel, T. Singer, A. R. Ofial and H. Mayr, Eur. J. Org. Chem., 2009, 3203–3211 CrossRef CAS.
- Phase transfer-catalyzed additions to p-QMs: (a) W.-D. Chu, L.-F. Zhang, X. Bao, X.-H. Zhao, C. Zeng, J.-Y. Du, G.-B. Zhang, F.-X. Wang, X.-Y. Ma and C.-A. Fan, Angew. Chem., Int. Ed., 2013, 52, 9229–9233 CrossRef CAS PubMed; (b) L. Ge, X. Lu, C. Cheng, J. Chen, W. Cao, X. Wu and G. Zhao, J. Org. Chem., 2016, 81, 9315–9325 CrossRef CAS PubMed; (c) Y.-J. Fan, L. Zhou and S. Li, Org. Chem. Front., 2018, 5, 1820–1824 RSC.
- Addition of masked α-amino acid derivatives to p-QMs: (a) X.-Z. Zhang, Y.-H. Deng, X. Yan, K.-Y. Yu, F.-X. Wang, X.-Y. Ma and C.-A. Fan, J. Org. Chem., 2016, 81, 5655–5662 CrossRef CAS PubMed; (b) F.-S. He, J.-H. Jin, Z.-T. Yang, X. Yu, J. S. Fossey and W.-P. Deng, ACS Catal., 2016, 6, 652–656 CrossRef CAS; (c) W. Li, X. Xu, Y. Liu, H. Gao, Y. Cheng and P. Li, Org. Lett., 2018, 20, 1142–1145 CrossRef CAS PubMed; (d) Z.-P. Zhang, K.-X. Xie, C. Yang, M. Li and X. Li, J. Org. Chem., 2018, 83, 364–373 CrossRef CAS PubMed.
- For further details see the online ESI†.
- (a) T. Ooi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 1999, 121, 6519–6520 CrossRef CAS; (b) T. Ooi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 2003, 125, 5139–5151 CrossRef CAS PubMed.
- Asymmetric PTC reactions using 0.01 mol% of a chiral PTC: (a) M. Kitamura, S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2005, 44, 1549–1551 CrossRef CAS PubMed; (b) Z. Han, Y. Yamaguchi, M. Kitamura and K. Maruoka, Tetrahedron Lett., 2005, 46, 8555–8558 CrossRef CAS; (c) M. Kitamura, S. Shirakawa, Y. Arimura, X. Wang and K. Maruoka, Chem. – Asian J., 2008, 3, 1702–1714 CrossRef CAS PubMed.
- CCDC 1911984 (http://www.ccdc.cam.ac.uk) contains the crystallographic data for 3j and CCDC 1943927 those for 3b†.
- For the intermediate formation of a similar CF3-p-QM: Y. Gong and K. Kato, Synlett, 2002, 431–434 CrossRef.
- (a) J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS PubMed; (b) T. G. Wucherpfennig, V. R. Pattabiraman, F. R. P. Limberg, J. Ruiz-Rodriguez and J. W. Bode, Angew. Chem., Int. Ed., 2014, 53, 12248–12252 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, analytical details, copies of NMR spectra, HPLC traces and X-ray crystallographic data. CCDC 1911984 (3j) and 1943927 (3b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc09239k |
| This journal is © The Royal Society of Chemistry 2020 |