
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic thin film photofaradaic pixels for on-demand electrochemistry in physiological conditions†
Maciej
Gryszel
 ab and
Eric Daniel
Głowacki
*abc
ab and
Eric Daniel
Głowacki
*abc
aLaboratory of Organic Electronics, ITN Campus Norrköping, Linköping University, Norrköping, Sweden. E-mail: eric.glowacki@liu.se
bWallenberg Centre for Molecular Medicine (WCMM) Linköping University, Linköping, Sweden
cWarsaw University of Technology, Faculty of Chemistry, Warsaw, Poland
First published on 2nd January 2020
Abstract
We report ultrathin organic photovoltaic elements optimized to run photofaradaic reactions in biological conditions. We demonstrate concurrent oxygen reduction to hydrogen peroxide and glucose oxidation. The devices are powered by deep-red irradiation in the tissue transparency window. We utilize bilayers of phthalocyanine, acting as the light absorber, and perylene diimide, functioning as both electron-acceptor and the hydrogen peroxide evolution electrocatalyst. These heterojunction bilayers are stable when irradiated in simulated physiological conditions, producing photovoltages sufficient to simultaneously drive cathodic oxygen reduction to H2O2 and anodic oxidation of glucose. We find that optimization of the anode metal is critical for sustained photofaradaic reactivity. Our results demonstrate a robust “wet” thin film photovoltaic with potential for physiological applications where localized electrochemical manipulation is desired, in particular the delivery of reactive oxygen species.
Light-triggered production of reactive oxygen species (ROS) is central to photodynamic therapy, where generation of singlet oxygen is desired.1 While delivery of 1O2 is well-established, recently the ROS species H2O2 has attracted attention as a signaling molecule effecting various (electro)physiological processes.2–4 Levels of H2O2 in the micromolar range, for instance, can regulate the gating of voltage-gated potassium channels which are critical to the behavior of excitable cells.5 Manipulation of local H2O2 also can be used to affect mitochondrial metabolic processes. The use of organic semiconducting nanoparticles as light absorbers to photogenerate ROS has already begun to gain traction in biophysical research.6–8 In our own research we have introduced aqueous organic dyes for photochemical ROS generation.9 The aim of this work is to develop a solid-state photofaradaic device which can produce H2O2via O2 reduction, while consuming sacrificial electron donors available in physiological conditions. This device should operate in the tissue transparency window (roughly 620–800 nm). The great body of work on photocatalytic generation of H2O2 in aqueous conditions relies on higher energy photons.10 For this reason, here we use an organic heterojunction with broad absorption throughout the deep-red region. The metal-free phthalocyanine/N,N′-dimethylperylenetetracarboxylic diimide (H2Pc/PTCDI) materials system we have elaborated previously in our studies of organic electrolytic photocapacitors (OEPCs),11,12 photodiodes for the driving of electrophoretic ion pumps,13 and finally as photocathodes for O2 reduction to H2O2.14 A salient feature emerging from these works is the remarkable stability of the H2Pc/PTCDI donor/acceptor (PN) junction.12 OEPC devices with a H2Pc/PTCDI heterojunction operate in AC mode with short light impulses. Initial tests of long-term pulsing in artificial cerebrospinal fluid demonstrated that hydrogen peroxide forms in the solution.12 Peroxide formation was only observed when the back contact of the device was gold. In the case where the back contact used was indium tin oxide (ITO), no detectable levels of peroxide were found. This can be rationalized by the fact that gold can support various faradaic reactions,15 while ITO has a large inert potential window.16 This finding points out that for net peroxide production, a photofaradaic cycle with a cathodic and anodic component must be supported. Here we detail how stable, DC photofaradaic devices can be produced for continuous H2O2 generation in biological environments.
We fabricated PN bilayers (30 + 30 nm) on semitransparent, evaporated gold layers (10 nm thickness). As a substrate for this and subsequent tests, we used Parylene-C. Parylene-C was chosen since it is a well-established ultrathin polymeric material for biocompatible implants.17 The PN bilayer serves as the charge generation layer: photogenerated excitons separate at the donor/acceptor interface, transfering electrons to the PTCDI layer. PTCDI is an established electrocatalyst for O2 reduction to H2O2,18 and this PN charge generation layer we have previously optimized for photocathodes for H2O2 generation.14 Photogenerated holes, meanwhile, are injected into the gold layer, where they can lead to oxidation of suitable electron donors in solution. For relevance to implantable devices in biomedical applications aimed at interfacing with the nervous system, we conducted experiments in artificial cerebrospinal fluid (aCSF). Following continuous irradiation with an LED emitting at 660 nm for 30 min, a concentration of 130 μm H2O2 was measured in the solution. Pictures of the experimental setup can be found in Fig. S1 (ESI†). Repeating this experiment for several more 30 min cycles (same device, fresh electrolyte) led to rapid decrease in the amount of evolved H2O2 (Fig. 1b). We hypothesized that unfortunately the PN layers may be unstable under conditions of continuous high-intensity irradiation. We measured the UV-Vis spectrum of the used device, finding that in fact that spectrum was unchanged with respect to an as-fabricated device (Fig. 1c). Moreover, we registered the photovoltage using the transient electrophotoresponse method,11 and found that the photovoltage and charging dynamics of the PN device were unchanged after the repeated runs (Fig. 1d). Therefore, the PN component itself appears to be stable and the origin of the H2O2 production drop-off must be something else. We suspected that the Au back contact may be the source of the problem. From work on OEPCs,12 we knew that the back contact is critical to the overall capacitive versus faradaic performance of the device. We verified first that the Au layer was not degraded and of lower conductivity, it in fact had the same resistance as measured with a multimeter before and after. It led us to the hypothesis that the Au layer had, at the microscopic level, lost its electrocatalytic properties. As a rapid empirical evidence that this may be the case, we evaporated a 10 nm layer of new gold on top of the part of the sample which was not covered by the PN junction, and measured a nearly threefold increase in the evolved H2O2 (Fig. 1b). This performance again declined with more subsequent runs – the Au was losing its catalytic properties. It was therefore clear that the PN layers provided stable operation, but faradaic performance of the gold back contact was the limitation to sustained faradaic behavior. Considering the photovoltage produced by the Au/PN device and the composition of the aCSF, it is clear that the only possible oxidizable species is glucose. We proceeded thus with the understanding that the back-contact material must be optimized with respect to stability as a glucose oxidizing electrocatalyst.
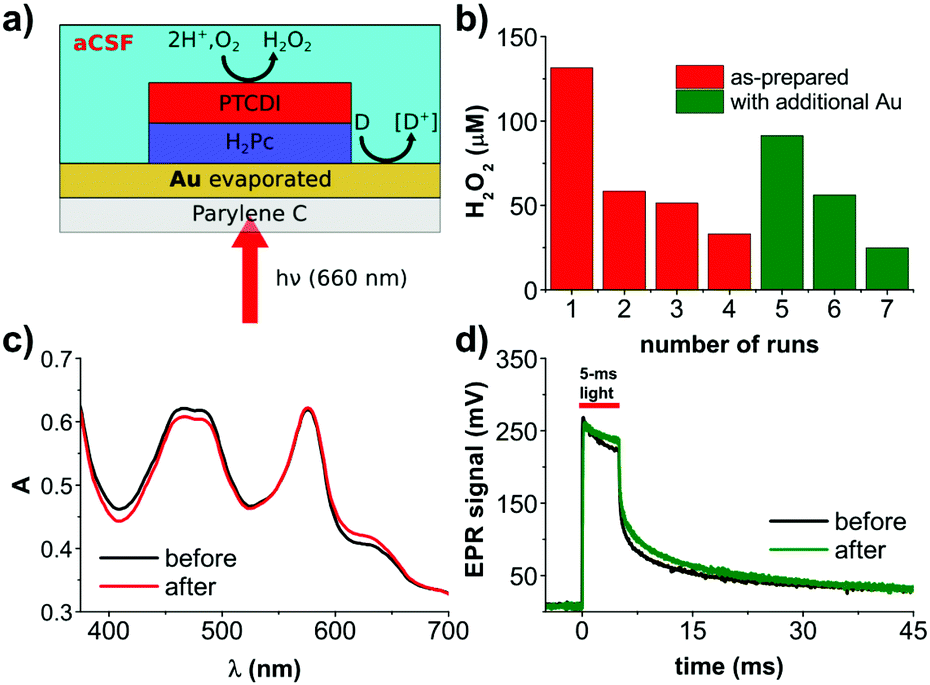 | ||
| Fig. 1 Organic photofaradaic devices for H2O2 evolution. (a) Device structure schematic, comprising a Parylene-C 5 μm-thick foil, semitransparent layer of gold (10 nm, evaporated) and an organic PN junction, fabricated from organic pigments: H2Pc (P type) and PTCDI (N type). Devices are fully immersed in aCSF electrolyte and irradiated with 660 nm light. (b) H2O2 concentrations measured in the electrolyte around the device in oxygen-containing aCSF solution under constant irradiation with red light (660 nm, 11 mW mm−2). The chart presents final [H2O2] at the end of each 30 min experiment. After each cycle, the electrolyte has been replaced with fresh solution. After the 4th experiment, the external layer of gold has been refreshed by thermal evaporation of additional 10 nm of Au. This series of experiments shows the crucial role of gold activity for the performance of the system. (c) Comparison of the UV-Vis spectra of the PN system, before and after the 4th experiment shows its photostability. (d) Electrical photoresponse of the PN system before and after the 4th experiment shows stability of the photogenerated voltage. | ||
We therefore focused our attention on exploring potential electrocatalysts for glucose oxidation. Many examples exist in the literature concerning metallic catalysts based on Cu, Ag, Pd, Pt, or enzymes.19 In the case of our application, the catalyst must not only be highly stable in a chloride-containing electrolyte, but also should not catalyze the decomposition of or otherwise react with hydrogen peroxide. Cu or Ag do not have sufficient electrochemical stability to work in our device in the given conditions, while materials like Pt rapidly decompose H2O2. We tested evaporated Pd films and found that they too decompose H2O2. This led us to dig deeper into optimizing gold itself as a glucose oxidation catalyst. The nuances of glucose oxidation on gold surfaces are well-described in the literature, especially in relation to nonenzymatic amperometric glucose sensors.19 In many of such studies, gold is electrodeposited on top of an underlying conductor. We elected to pursue the same approach, and try electrodeposited gold layers around the PN pixel on preevaporated gold thin films. The thin evaporated gold under the PN pixel is necessary to maintain semitransparency. We attempted different gold electrodeposition conditions (Fig. S2, ESI†), finding favorable conditions using HAuCl4 as the precursor20 and two-electrode potentiostatic deposition at −2 V versus the counter electrode. A comparison of results from different electrodeposition conditions, as well different evaporated and sputtered Au films, can be found in Fig. S2 (ESI†). Fig. 2a illustrates this photofaradaic pixel modified with the optimal ring of electrodeposited gold. The performance of these modified devices was significantly higher in terms of peroxide yield, and the stability was likewise much better. In total, thirteen rounds of 30 minute irradiation experiments were performed, showing the enhanced performance stability afforded by the electrodeposited gold. After these repeated cycles, the UV-Vis absorption spectrum and the EPR photovoltage were recorded (Fig. 2c and d) and once again proved that the organic PN junction layers were undegraded. Scanning electron microscopy (SEM) of PN layers before and after the experiment likewise showed no significant changes in morphology, pinhole formation, etc. (Fig. 2d). Encouraged by the better performance afforded by electrodeposited gold layers, we tested a longer irradiation experiment, 180 min, and found that H2O2 rises to around 2000 μM, at which point it saturates. Such a peroxide saturation concentration can be rationalized considering the competitive process of H2O2 being further reduced to H2O. This process begins to become efficient when the [H2O2] and [O2] start to reach similar levels in the solution. In O2 saturated solutions, where [O2] is around 1–2 mM, PTCDI as an electrocatalyst starts leveling off at an H2O2 equilibrium concentration of 20–30 mM.18 In ambient air as in the experiments done here, the [O2] is around 200–300 μM, therefore a saturated H2O2 concentration of 2 mM is reasonable. It should also be considered that glucose is present at 10 mM concentration, therefore by the end of this experiment it may also become a limiting reagent. Finally, the stability of H2O2 should be taken into account. We measured a background decomposition rate of roughly 500 nM min−1 under irradiation of the blank electrolyte alone. This rate was found not to be changed by presence of Au.
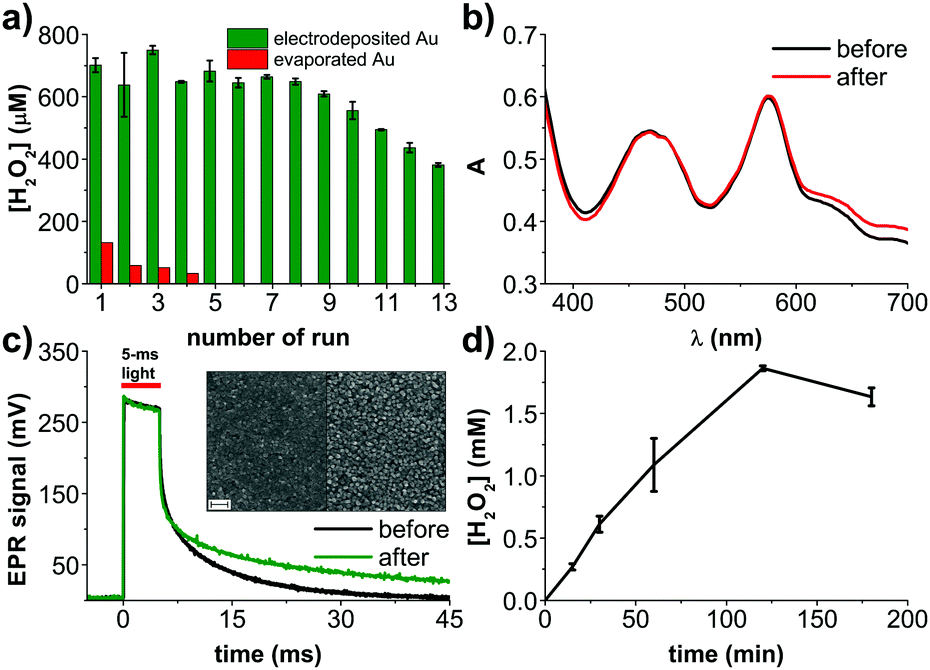 | ||
| Fig. 2 Devices with electrodeposited Au anodes. (a) [H2O2] produced by the device in oxygen-containing aCSF solution under constant irradiation with red light (660 nm, 11 mW mm−2). The chart presents final [H2O2] at the end of each, 30 min cycle. Compared with PN devices with evaporated Au, it shows not only much higher performance but also higher stability. (b) Comparison of the UV-Vis spectra of the PN system, before and after the 13th experiment. The PN layer is photostable. (c) Electrical photoresponse of the PN system before and after the 13th experiment, evidencing no decrease of the generated photovoltage. (c, inset) Comparison of SEM images of the PN structure before and after the 13th experiment. Shape and average size of the nanocrystalline PTCDI domains remains unchanged. Scale bar = 200 nm. (d) H2O2 photoevolution over 180 min, showing saturation of [H2O2] after 2 h. | ||
To better understand the role of the Au anode component of the photofaradaic pixel, we performed electrochemical measurements on the gold films alone. From CV scans of comparing electrodeposited Au films in aCSF with and without glucose, oxidation peaks resulting from the addition of glucose are clearly visible (Fig. 3a). In contrast, evaporated Au films are a poor electrocatalytic surface. The CV (Fig. 3b) shows roughly ten times lower anodic current, much less apparent peaks, and lower capacitive current on the backward scan when glucose is included in the solution, all together suggesting rapid poisoning of the catalytic surface even during the course of a single cycle. The poisoning of Au surfaces due to glucose oxidation is described in the literature.21 We tested stability and performance by performing chronoamperometry on the Au electrodes (Fig. 3c). Here again the much higher and sustained electrocatalytic performance of electrodeposited versus evaporated films is evidenced. A primary cause would appear to be simply the much higher electrochemical surface area of the electrodeposited gold films, apparent in SEM images (Fig. 3d).
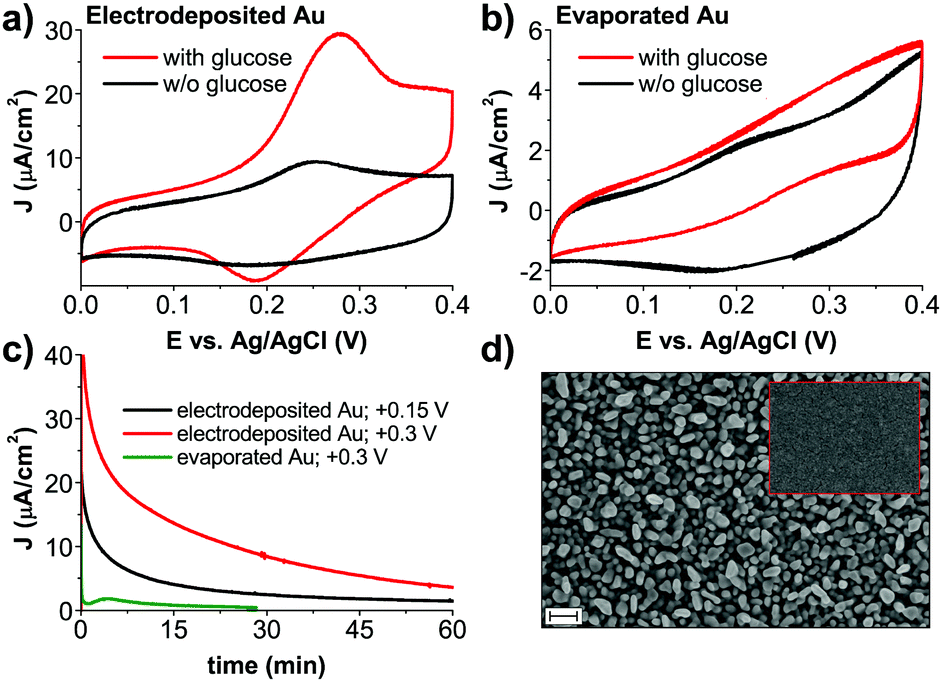 | ||
| Fig. 3 Electrochemical characterization of different types of gold used for glucose oxidation. ((a) electrodeposited; and (b) evaporated) Comparison of CVs, obtained in aCSF with and without glucose. Glucose oxidation is clearly visible, with behavior in glucose-free conditions corresponding to the redox peak of Au in chloride-containing electrolyte.22 (c) Chronoamperometric stability test for aCSF with glucose. The solution has not been deoxygenated for consistency with conditions of photochemical H2O2 evolution. All values of applied potentials are given versus Ag/AgCl reference electrode (d) SEM image of electrodeposited Au before H2O2 photoevolution. During the course of the experiment morphology of gold does not change. For comparison, the inset shows a SEM of an evaporated gold electrode. Scale bar = 200 nm for both images. | ||
Promising results with the improved back contact encouraged us to seek optimization of the cathodic side as well. From our earlier work on photocathodes for oxygen reduction we knew that electron-accepting Au contacts topped with an additional thin layer of PTCDI can increase photocathodic current significantly; or that vertical PN/PN tandems can boost performance as well.14 We hypothesized that these improvements would lead to higher photovoltages to provide greater overpotential to increase glucose oxidation. EPR measurements of improved PN structures confirm that voltage can be increased (Fig. S4a, ESI†). Irradiating these different samples showed that the structures optimized for higher voltage produce less H2O2 than the basic PN (Fig. S4b, ESI†). This can only be the case if the additional voltage is in fact resulting in reduction of O2 all the way to H2O, resulting in lower [H2O2]. This underscores an important aspect of these types of photofaradaic devices, which are electrically floating. One must consider to balance the anodic and cathodic performance to obtain the desired net faradaic reaction.
The result of our efforts is a free-standing photofaradaic unit on a thin plastic substrate, which demonstrates robustness in on-demand H2O2 generation. We have engineered the device in the direction of eventual biomedical devices operating in physiological conditions, focusing on glucose as a target electron donor. A critical message of our work is that that an electrically-floating photovoltaic device must balance cathodic and anodic reactions to give the net sustained reaction. Simultaneous ROS production with local oxidation of glucose as the electron donor suggests this type of platform for combination chemodynamic/starvation therapy, an emerging targeted cancer treatment approach.23 Future work should focus on evaluation of devices in biological in vitro and in vivo conditions, where it will be deployed for localized H2O2 delivery. This approach with light-actuation can enable many previously impossible experiments and eventually inspire new therapeutic directions as well.
The authors gratefully acknowledge financial support from the Knut and Alice Wallenberg Foundation, within the framework of the Wallenberg Centre for Molecular Medicine at Linköping University, as well as the Wallenberg Wood Science Centre 2.0; and the Swedish Research Council (Vetenskapsrådet, 2018-04505).
Conflicts of interest
There are no conflicts to declare.Notes and references
- Photodynamic Therapy Methods and Protocols, ed. C. J. Gomer, Humana Press, New York, 1998 Search PubMed.
- J. R. Stone and S. Yang, Antioxidants Redox Signal., 2006, 8, 243–270 CrossRef PubMed.
- B. C. Dickinson and C. J. Chang, Nat. Chem. Biol., 2011, 7, 504–511 CrossRef PubMed.
- R. Bretón-Romero and S. Lamas, Redox Biol., 2014, 2, 529–534 CrossRef PubMed.
- N. Gamper, O. Zaika, Y. Li, P. Martin, C. C. Hernandez, M. R. Perez, A. Y. C. Wang, D. B. Jaffe and M. S. Shapiro, EMBO J., 2006, 25, 4996–5004 CrossRef PubMed.
- M. Moros, A. Lewinska, G. Onorato, M. R. Antognazza, F. Di Maria, M. Blasio, G. Lanzani, A. Tino, M. Wnuk and C. Tortiglione, MRS Commun., 2018, 1–8 Search PubMed.
- M. R. Antognazza, I. A. Aziz and F. Lodola, Oxid. Med. Cell. Longev., 2019, 1–24 Search PubMed.
- F. Lodola, V. Rosti, G. Tullii, A. Desii, L. Tapella, P. Catarsi, D. Lim, F. Moccia and M. R. Antognazza, Sci. Adv., 2019, 5, eaav4620 CrossRef.
- M. Gryszel, R. Rybakiewicz and E. D. Głowacki, Adv. Sustainable Syst., 2019, 3, 1900027 CrossRef.
- S. Fukuzumi, Y. M. Lee and W. Nam, Chem. – Eur. J., 2018, 24, 5016–5031 CrossRef.
- D. Rand, M. Jakešová, G. Lubin, I. Vebraite, M. David-Pur, V. Đerek, T. Cramer, N. S. Sariciftci, Y. Hanein and E. D. Głowacki, Adv. Mater., 2018, 30, 1707292 CrossRef.
- M. Jakešová, M. S. Ejneby, V. Đerek, T. Schmidt, M. Gryszel, J. Brask, R. Schindl, D. T. Simon, M. Berggren, F. Elinder and E. D. Głowacki, Sci. Adv., 2019, 5, eaav5265 CrossRef.
- M. Jakešová, T. A. Sjöström, V. Đerek, D. Poxson, M. Berggren, E. D. Głowacki and D. T. Simon, npj Flex. Electron., 2019, 3, 14 CrossRef.
- M. Gryszel, A. Markov, M. Vagin and E. D. Głowacki, J. Mater. Chem. A, 2018, 6, 24709–24716 RSC.
- L. D. Burke and P. F. Nugent, Gold Bull., 1998, 31, 39–50 CrossRef.
- J. D. Benck, B. A. Pinaud, Y. Gorlin and T. F. Jaramillo, PLoS One, 2014, 9, e107942 CrossRef.
- J. B. Fortin and T.-M. Lu, Chemical Vapor Polymerization, The Growth and Properties of Parylene, Springer Science + Business Media, New York, 1st edn, 2004 Search PubMed.
- M. Warczak, M. Gryszel, M. Jakešová, V. Đerek and E. D. Głowacki, Chem. Commun., 2018, 54, 1960–1963 RSC.
- A. Brouzgou and P. Tsiakaras, Top. Catal., 2015, 58, 1311–1327 CrossRef.
- T. M. Cheng, T. K. Huang, H. K. Lin, S. P. Tung, Y. L. Chen, C. Y. Lee and H. T. Chiu, ACS Appl. Mater. Interfaces, 2010, 2, 2773–2780 CrossRef.
- M. Tominaga, M. Nagashima, K. Nishiyama and I. Taniguchi, Electrochem. Commun., 2007, 9, 1892–1898 CrossRef.
- L. D. Burke and P. F. Nugent, Gold Bull., 1997, 30, 43–53 CrossRef.
- M. Chang, M. Wang, M. Wang, M. Shu, B. Ding, C. Li, M. Pang, S. Cui, Z. Hou and J. Lin, Adv. Mater., 2019, 31, 1905271 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methods. See DOI: 10.1039/c9cc09215c |
| This journal is © The Royal Society of Chemistry 2020 |