
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethylation of geometrically constrained lysine analogues by histone lysine methyltransferases†
Abbas H. K.
Al Temimi
 a,
Paul B.
White
a,
Marcus J. M.
Mulders
b,
Nicole G. A.
van der Linden
a,
Richard H.
Blaauw
c,
Anita
Wegert
*b,
Floris P. J. T.
Rutjes
*a and
Jasmin
Mecinović
*ad
a,
Paul B.
White
a,
Marcus J. M.
Mulders
b,
Nicole G. A.
van der Linden
a,
Richard H.
Blaauw
c,
Anita
Wegert
*b,
Floris P. J. T.
Rutjes
*a and
Jasmin
Mecinović
*ad
aInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: Floris.Rutjes@ru.nl; Tel: +31 24 3653202
bMercachem B.V., Kerkenbos 1013, 6546 BB Nijmegen, The Netherlands. E-mail: anita.wegert@mercachem.nl; Tel: +31 24 3652331
cChiralix B.V., Kerkenbos 1013, 6546 BB Nijmegen, The Netherlands
dUniversity of Southern Denmark, Campusvej 55, 5230 Odense, Denmark. E-mail: mecinovic@sdu.dk; Tel: +45 65 503603
First published on 3rd February 2020
Abstract
We report synthesis and enzymatic assays on human histone lysine methyltransferase catalysed methylation of histones that possess lysine and its geometrically constrained analogues containing rigid (E)-alkene (KE), (Z)-alkene (KZ) and alkyne (Kyne) moieties. Methyltransferases G9a and GLP do have a capacity to catalyse methylation in the order K ≫ KE > KZ ∼ Kyne, whereas monomethyltransferase SETD8 catalyses only methylation of K and KE.
Histone lysine methylation is a universal posttranslational modification that is engaged in the regulation of many biological processes, and its dysregulation is linked to various diseases, including cancer, inflammation, and genetic disorders.1 Histone lysine methyltransferases (KMTs) in the presence of cosubstrate S-adenosylmethionine (SAM) catalyse methylation of lysine residues in histones and non-histone proteins via addition of one, two or three methyl groups (Kme, Kme2 and Kme3) on the ε-amino group of the target lysine (Fig. 1a).2,3 Demethylation of Nε-methyllysine is catalysed by the flavin-dependent and the 2-oxoglutarate/Fe(II) dependent demethylases (KDMs),4 while different aromatic cage containing epigenetic reader proteins specifically recognise histone Nε-methyllysine residues.5 Recent biostructural studies on ternary complexes between KMTs, histone peptides and S-adenosylhomocysteine (SAH, as a non-reactive product of the methyl transfer reaction) revealed that the SET (Su(var)3-9, enhancer-of-zeste, trithorax) domain containing KMTs have a well-defined arrangement of the binding pockets for the lysine residue and SAM, which are located at opposite sides of the SET domain and connected through a narrow hydrophobic tunnel (Fig. 1b).6 Mutation studies demonstrated that the nature of specific residues (Tyr and Phe) in the active site of the SET domain apparently determines the methylation level of particular KMTs. The Tyr OH group interacts with the ε-amine of lysine by a favourable NH⋯O hydrogen bond, thus orienting the amino group toward the methyl moiety of SAM for efficient methyl transfer. Kinetic studies reveled that the KMT-catalysed methyl transfer from SAM to the target lysine proceeds via an early asymmetrical SN2 transition state.7 The biocatalytic potential of KMTs has remained largely undetermined, despite significant progress in mechanistic, structural and inhibition studies on histone KMTs.6,8
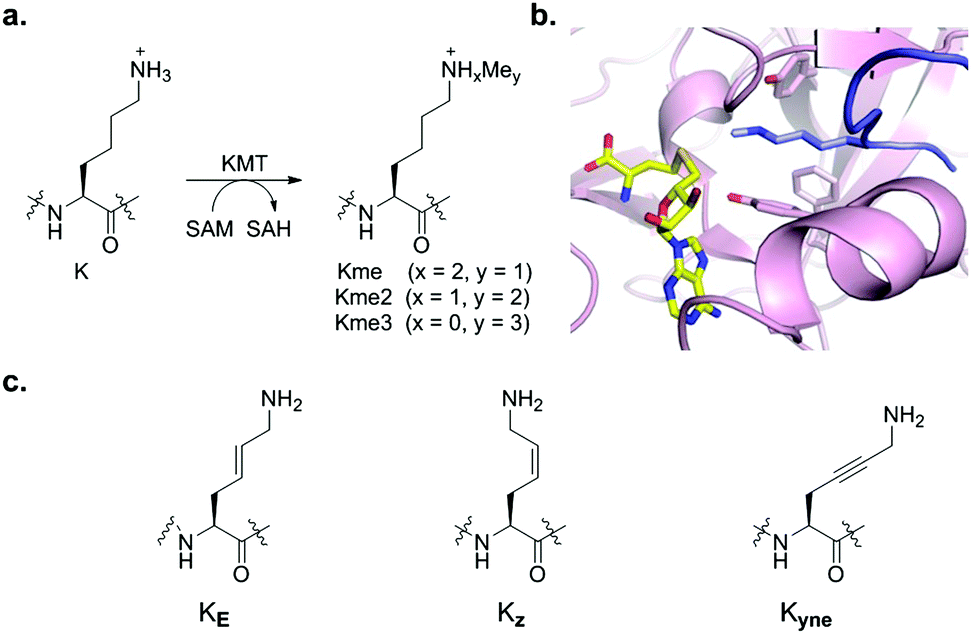 | ||
| Fig. 1 (a) KMT-catalysed methylation of lysine residues in histones. (b) View from a crystal structure of GLP (magenta) in complex with H3K9me histone peptide (blue) and S-adenosylhomocysteine (SAH, yellow). (c) Geometrically constrained lysine analogues as substrates for KMTs. | ||
Recent studies have expanded the knowledge concerning the biocatalytic activity of human KMTs via examining various lysine analogues as substrates, revealing that KMTs not only display a high degree of specificity towards lysine, but also to simple lysine analogues.9–16 Despite the progress in understanding of the role of the substrate-binding pocket on KMT catalysis, it is presently unclear how does the flexible lysine side chain contributes to substrate efficiency. In the ternary complex, the histone lysine side chain typically exhibits the anti (zig-zag) conformation, presumably enabling an optimal positioning towards the SAM cosubstrate for efficient methyl transfer (Fig. 1b). In this communication, we explore the importance of the conformational freedom of C–C bonds in the lysine side chain by investigating whether KMTs do have a capacity to catalyse methylation of the simplest geometrically constrained lysine analogues incorporated in histone peptides.
We hypothesised that the conformational freedom of the lysine side chain may have a major influence on the catalytic activity of KMTs. To explore the significance of conformational restriction, we introduced different rigidifying elements at the lysine side chain having the same number of core atoms, but less conformational freedom. The three selected analogues are: (i) KE, an unsaturated lysine having an (E)-configured double bond between the γ- and δ-positions,17 (ii) KZ, an unsaturated lysine having a (Z)-configured double bond between the γ- and δ-positions,18,19 and (iii) Kyne, an unsaturated lysine having a triple bond between the γ- and δ-positions (Fig. 1c).20,21 Experimental details for the preparation of the three Fmoc-protected building blocks, proof of purity, and NMR data for their characterization are elaborated in the ESI.† The structures and syntheses of Fmoc-protected lysine analogues are shown in Scheme 1a. 1H NMR and analytical SFC studies confirmed the identical structures of compound 2 obtained either via cross-metathesis or via flow hydrogenation of 3 (Fig. S5 and S6, ESI†). 1D selective NOESY experiments were employed to confirm the alkene geometry of KZ and KE (Fig. S4, ESI†).
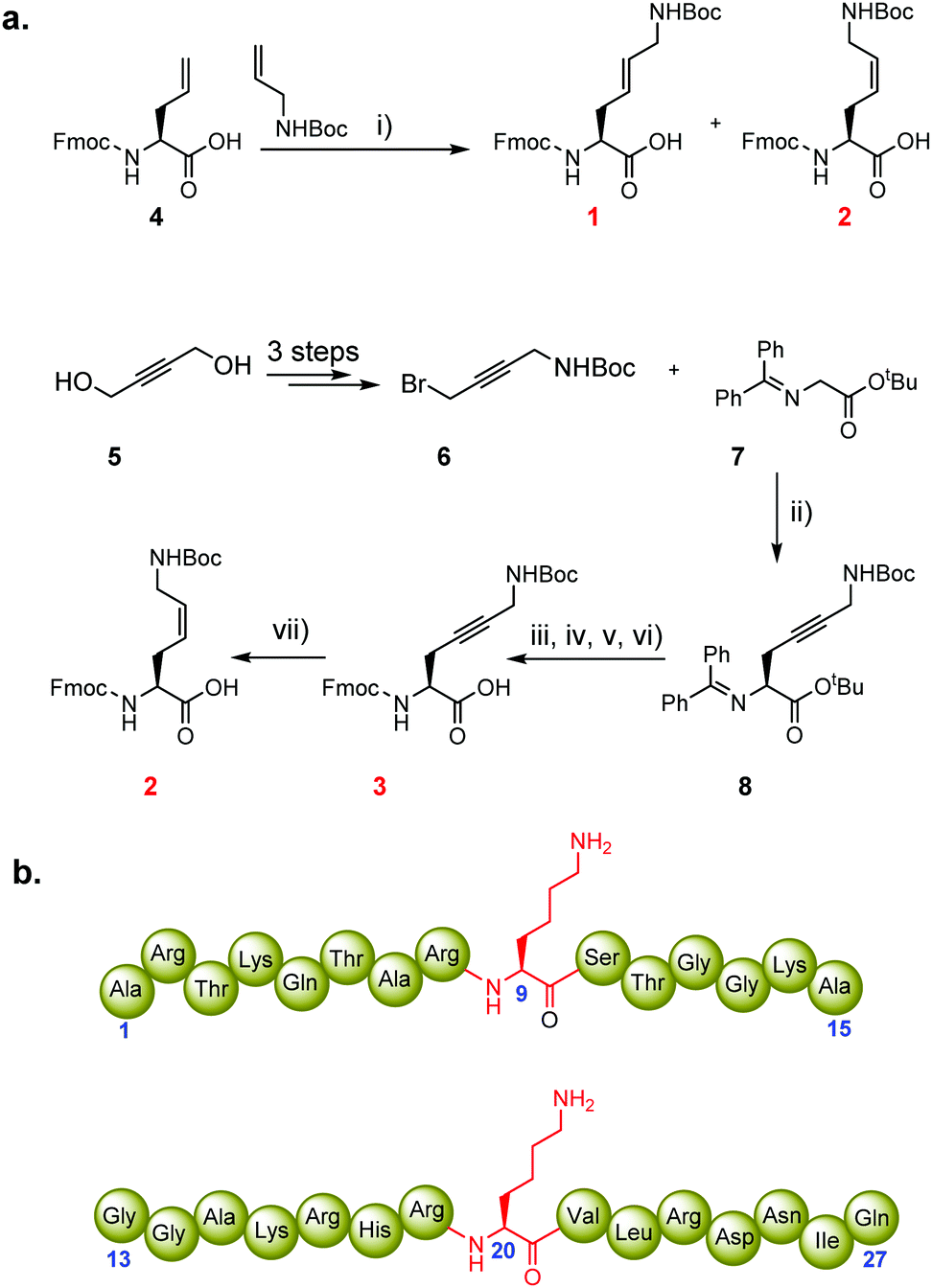 | ||
| Scheme 1 Synthesis of Fmoc-protected conformationally restricted lysine analogues for Fmoc-SPPS, and sequences of synthetic histone peptides. (a) Reagents and conditions: (i) 10 mol% HGII, CH2Cl2, reflux, 16 h, 37% (1, KE) and 3% (2, KZ); (ii) 10 mol% Corey's catalyst, CsOH·H2O, CH2Cl2, −78 °C, 24 h, 45%; (iii) citric acid (15% aq), THF, 0 °C → rt, 83%; (iv) Fmoc-OSu, DIPEA, CH2Cl2, 82%: (v) TFA, 3 h, rt; (vi) Boc2O, DIPEA, MeCN, 16 h, rt, 57%; (vii) H-Cube hydrogenation, Lindlar catalyst, EtOAc, 10 °C, 38% (2, KZ). HGII = Hoveyda–Grubbs 2nd generation catalyst; Fmoc-OSu = Fmoc N-hydroxysuccinimide ester; DIPEA = diisopropylethylamine; Boc = tert-butoxycarbonyl. (b) Representative structures of histone H3K9 (1–15 aa, top) and H4K20 (13–27 aa, bottom) peptides. | ||
To explore the substrate specificity of the geometrically constrained lysine analogues by human KMTs, we synthesised histone peptides corresponding to the sequence of H3 and H4 histones and subjected them to methylation by recombinantly expressed human di-/trimethyltransferases GLP and G9a, and monomethyltransferase SETD8. The chemically synthesised histone peptides are shown in Scheme 1b (H3 residues 1–15, ARTKQTAR![[X with combining low line]](https://www.rsc.org/images/entities/char_0058_0332.gif) 9STGGKA; H4 residues 13–27, GGAKRHR20VLRDNIQ; Schemes S1, S2 and Fig. S7, S8, ESI†). Using the three Fmoc-protected lysine analogues 1–3, we constructed six peptides substituted at the indicated position with the lysine analogue (position 9 of H3, position 20 of H4). All histone peptides were produced by SPPS, purified by reversed-phase HPLC, and analysed by analytical HPLC, LC-MS and MALDI-TOF MS (Fig. S9–S14, ESI†).
9STGGKA; H4 residues 13–27, GGAKRHR20VLRDNIQ; Schemes S1, S2 and Fig. S7, S8, ESI†). Using the three Fmoc-protected lysine analogues 1–3, we constructed six peptides substituted at the indicated position with the lysine analogue (position 9 of H3, position 20 of H4). All histone peptides were produced by SPPS, purified by reversed-phase HPLC, and analysed by analytical HPLC, LC-MS and MALDI-TOF MS (Fig. S9–S14, ESI†).
To identify new substrates from these histone peptides, we first performed enzymatic assays of KMT-catalysed methylation and analysed the formation of methylated products by MALDI-TOF MS. Standard assays were carried out in the presence of the histone peptide (100 μM), enzyme (2 μM), SAM (500 μM for GLP and G9a; 200 μM for SETD8) in Tris–HCl buffer at pH 8.0. After 1 hour incubation at 37 °C, MALDI-TOF MS data showed complete trimethylation of H3K9 in the presence of GLP and G9a, and almost complete monomethylation of H4K20 in the presence of SETD8 (Fig. 2a, e, and Fig. S15a, ESI†), in line with recent studies.9–13 Under the same conditions, the other six histone peptides containing the unsaturated KE, KZ, and Kyne lysine analogues were examined.
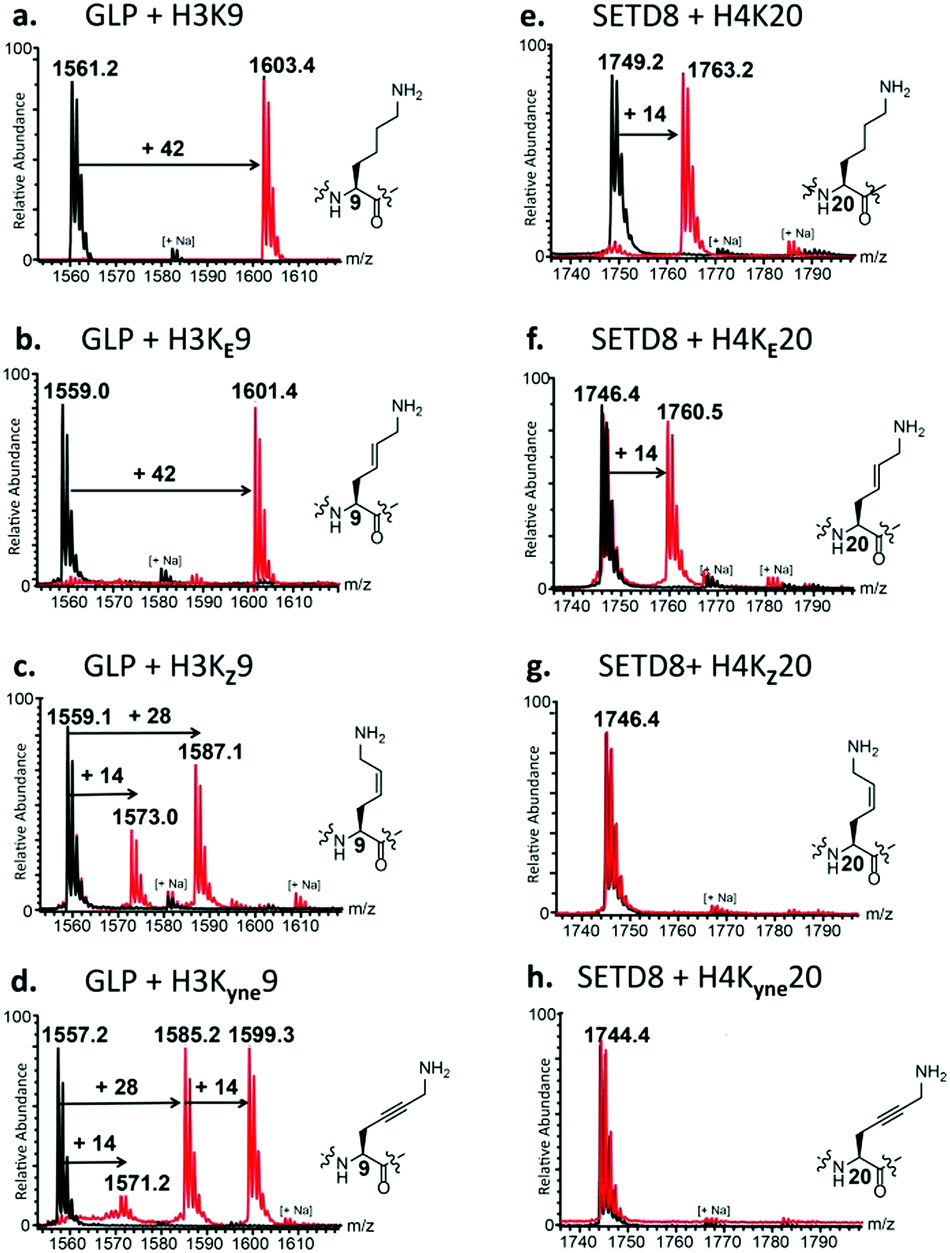 | ||
| Fig. 2 KMT-catalysed methylation of lysine and its conformationally restricted analogues. MALDI-TOF MS data showing methylation of the natural and unnatural histone peptides in the presence of GLP (left panels) with (a) H3K9, (b) H3KE9, (c) H3KZ9 and (d) H3Kyne9; and SETD8 (right panels) with (e) H4K20, (f) H4KE20, (g) H4KZ20, and (h) H4Kyne20. Red spectra show reactions in the presence of KMTs, while black spectra show the no-enzyme controls. | ||
First, we investigated the activity of H3KE9, H3KZ9, and H3Kyne9 with GLP and G9a. Strikingly, for both GLP and G9a we observed nearly complete trimethylation of H3KE9, producing H3KE9me3, similar to native H3K9 (Fig. 2b and Fig. S15b, ESI†). H3KZ9 underwent GLP-catalysed methylation to produce mainly H3KZ9me2, while a minor amount of H3KZ9me was also observed (Fig. 2c). Similarly, G9a catalysed a predominant dimethylation of H3KZ9 (Fig. S15c, ESI†). Prolonged incubation with GLP and G9a resulted in complete conversion into H3KZ9me2 and traces of H3KZ9me3 (Fig. S17, ESI†). Under standard conditions, both enzymes converted H3Kyne9 to H3Kyne9me2 and H3Kyne9me3, while only traces of H3Kyne9me were formed (Fig. 2d and Fig. S15d, ESI†). Prolonged incubation (3 h) with GLP and G9a led to complete conversion into H3Kyne9me3 (Fig. S16, ESI†). Control experiments in the absence of GLP/G9a with H3KE9, H3KZ9 and H3Kyne9 substrates verified that methylation reactions are due to the KMT activity (black spectra in Fig. 2). An increased concentration of G9a/GLP (4 μM) and SAM (1 mM) led to full trimethylation of H3KE9 and H3Kyne9, and mono- and dimethylated forms of H3KZ9 (Fig. S18, ESI†). Moreover, prolonged incubation using additional GLP (10 μM) and SAM (1 mM) led to significant amounts of H3KZ9me3 (Fig. S19, ESI†).
To further substantiate G9a/GLP-catalysed methylation of conformationally constrained lysine analogues, we carried out time course experiments (Fig. S20 and S21, ESI†). As anticipated at first glance and as described above, the data confirmed that GLP and G9a efficiently catalyse methylation of the three rigidified lysine analogues with the degree of methylation progressing over time. Time course data, however, also highlighted that different methylation states of lysine analogues were produced.
Enzyme kinetic assays in the presence of G9a revealed that lysine is a superior substrate over its geometrically constrained analogues; values of enzyme catalytic efficiency (kcat/KM) showed trends H3K9 ≫ H3KE9 > H3KZ9 ∼ H3Kyne9 (Table 1 and Fig. S22, ESI†). These differences arise from both higher KM and lower kcat values. Higher KM values might be a result of weaker binding that is possibly caused by a sterical clash between the geometrically constrained lysine analogue and the narrow lysine binding tunnel. We attribute lower kcat values to slower methyl transfer that is caused by non-optimal positioning of the nucleophilic amino group towards the electrophilic methyl group of SAM. It is noteworthy that the sp2/sp hybridised carbons in KE, KZ and Kyne lead to slightly shorter side chains with different spatial orientation when compared to the fully sp3 hybridised lysine side chain. It is thus very likely that the positioning of the ε-amino group in geometrically constrained lysine analogues does not allow the formation of the low energy collinear N⋯C⋯S transition state, resulting in slower methyl transfer reaction.
| Entry | H3 peptide | K M (μM) | k cat (min−1) | k cat/KM (mM−1 min−1) |
|---|---|---|---|---|
| 1 | H3K9 | 3.45 ± 0.6 | 20.7 ± 0.03 | 6012 |
| 2 | H3KE9 | 40.3 ± 7.3 | 13.0 ± 0.11 | 323 |
| 3 | H3KZ9 | 103 ± 48 | 4.39 ± 0.2 | 42.8 |
| 4 | H3Kyne9 | 158 ± 46 | 8.01 ± 0.22 | 50.6 |
We were then motivated to examine the analogues H4KE20, H4KZ20, and H4Kyne20 in the presence of human monomethyltransferase SETD8. Remarkably, SETD8 catalysed the conversion (50%) of H4KE20 to H4KE20me (Fig. 2f). Incubating the enzymatic mixture for 3 h at 37 °C afforded H4KE20me with nearly full conversion (Fig. S23, ESI†). In contrast, H4KZ20 and H4Kyne20 were not methylated within the limits of detection under the standard conditions (Fig. 2g and h). Even upon prolonged incubation with additional SETD8 (10 μM) and SAM (1 mM), methylation did not take place, indicating that SETD8 is highly specific for the trans geometrical isomer over the cis isomer (Fig. S24b and c, ESI†). Using the same high concentration of SETD8, almost full monomethylation of H4KE20 was observed in 1 h (Fig. S24a, ESI†).
Inhibition and residual activity assays monitoring KMT-catalysed methylation of the histone peptides were then carried out aimed at providing data on competitive binding of these three substrates as compared to the 14-mer H3K9 sequence to the active site of G9a/GLP. Among them, H3KE9 appeared the most potent competing substrate of GLP and G9a (Fig. S25, ESI†). H3KZ9 also showed significant competition with H3K9, while H3Kyne9 showed the lowest competition in this assay.
To further investigate the role of the rigidification of lysine side chain in GLP-catalysed methylation, we expanded our studies by carrying out 1H NMR and 1H–13C HSQC measurements. To verify that H3K9 undergoes GLP-catalysed methylation, we conducted enzymatic assays of the H3K9 peptide (400 μM) in the presence of GLP (8 μM) and SAM (2 mM) for 1 h at 37 °C in Tris–d11 buffer at pD 8.0. GLP catalysed trimethylation of H3K9, as illustrated by the appearance of a singlet at 3.03 ppm (13C 53.0 ppm, NMe3) (Fig. 3a and Fig. S32a, ESI†) and a triplet at 2.62 ppm (13C 28.0 ppm, SAH-CH2γ) due to concomitant formation of SAH, which is produced during the KMT-catalysed methylation of lysine and its simplest mimics.9,22 Non-methylated H3 histone peptides were also characterised by 1D and 2D NMR analyses (Fig. S26–S31, ESI†). Reactions with two substrates, H3KE9 and H3Kyne9, showed the appearance of new singlet resonances at 2.99 and 3.11 ppm, respectively (Fig. S32b and d, ESI†). Subsequent edited 1H–13C HSQC experiments revealed these signals as methyl groups with corresponding carbon resonances at 52.3 and 52.5 ppm, respectively (Fig. 3b and d). The downfield chemical shift of the 13C methyl groups conclusively identified these new resonances as part of an NMe3+ moiety, indicating that the lysine analogue was trimethylated, and matched the result obtained for H3K9. Further NMR spectroscopic analysis using 1H–13C HMBC and 1H–1H ROESY experiments confirmed the methylation of H3KE9 and H3Kyne9 occurred at the 9 position due to the long-range 1H–13C coupling between the trimethylammonium group and the Cε of the histone peptide, as well as long-range through-space interactions between the 1Hs of the trimethylammonium and the 1Hs of ε-CH2 (Fig. S33–S39, ESI†).
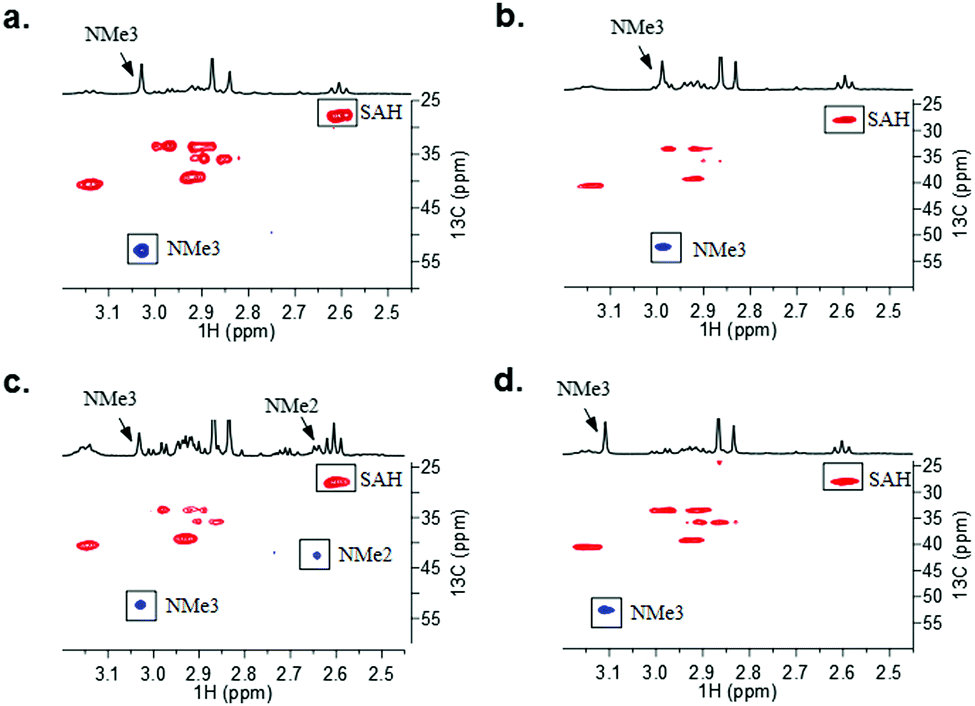 | ||
| Fig. 3 1H–13C HSQC data of GLP-catalysed methylation of (a) H3K9; (b) H3KE9; (c) H3KZ9; and (d) H3yne9 with assignment of cross-peaks. Correlations corresponding to the methylated lysine analogues are highlighted. | ||
H3KZ9 was unique among these analogues as two new singlet resonances appeared at 3.03 and 2.64 ppm in the 1H spectrum (Fig. 3c and Fig. S32c, ESI†). The singlet at 3.03 ppm was observed to have a 1-bond 1H–13C coupling to a 13C resonance at 52.5 ppm, similar to both H3KE9 and H3Kyne9, and was indicative of a trimethylated ammonium species, i.e. H3KZ9me3 (Fig. 3c). However, the other singlet observed at 2.64 ppm was directly coupled to a 13C resonance at 42.6 ppm. The upfield shift in both the 1H and 13C resonances (∼0.5 ppm for 1H and ∼10 ppm for 13C as compared to the other analogues) is consistent with dimethylation of H3KZ9 and literature reports of H3K9me2 and H3hK9me2. Results from GLP-catalysed methylation on H3KZ9 in the NMR assay were consistent with what was observed by MALDI-TOF MS data in the presence of increased amounts of GLP (Fig. S19, ESI†).
In conclusion, our results demonstrate that human KMTs bind the histone substrate containing an (E)-configured dehydrolysine (KE) in an orientation amenable for efficient methylation of the terminal Nε-amino group. We have also shown that the corresponding (Z)-configured dehydrolysine (KZ) and double unsaturated lysine (Kyne) mimics are novel substrates for GLP and G9a, while they are not methylated by SETD8, possibly due to their improper orientation for efficient methyl transfer. Overall, the work highlights that the conformational freedom of lysine side chain provides an important molecular and structural requirement for efficient KMT-catalysed methyl transfer reactions. We feel that these findings provide important fundamental molecular knowledge on the KMT enzyme family and may principally contribute to designing new chemical probes for KMTs.
We gratefully acknowledge financial support from the Dutch Research Council (NWO, NCI-TA grant 731.015.202) and European Research Council (ERC Starting Grant to J. M., ChemEpigen-715691). Dr Y. Vijayendar Reddy is acknowledged for fruitful discussions.
Conflicts of interest
There are no conflicts to declare.References
- T. Kouzarides, Cell, 2007, 128, 693–705 CrossRef CAS PubMed.
- J. C. Black, C. Van Rechem and J. R. Whetstine, Mol. Cell, 2012, 48, 491–507 CrossRef CAS PubMed.
- C. Martin and Y. Zhang, Nat. Rev. Mol. Cell Biol., 2005, 6, 838–849 CrossRef CAS PubMed.
- S. M. Kooistra and K. Helin, Nat. Rev. Mol. Cell Biol., 2012, 13, 297–311 CrossRef CAS.
- S. D. Taverna, H. Li, A. J. Ruthenburg, C. D. Allis and D. J. Patel, Nat. Struct. Mol. Biol., 2007, 14, 1025–1040 CrossRef CAS PubMed.
- M. Luo, Chem. Rev., 2018, 118, 6656–6705 CrossRef CAS PubMed.
- J. A. Linscott, K. Kapilashrami, Z. Wang, C. Senevirathne, I. R. Bothwell, G. Blum and M. Luo, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E8369–E8378 CrossRef CAS PubMed.
- M. Luo, ACS Chem. Biol., 2012, 7, 443–463 CrossRef CAS PubMed.
- A. H. K. Al Temimi, Y. V. Reddy, P. B. White, H. Guo, P. Qian and J. Mecinović, Sci. Rep., 2017, 7, 16148 CrossRef PubMed.
- R. Belle, A. H. K. Al Temimi, K. Kumar, B. J. G. E. Pieters, A. Tumber, J. E. Dunford, C. Johansson, U. Oppermann, T. Brown, C. J. Schofield, R. J. Hopkinson, R. S. Paton, A. Kawamura and J. Mecinović, Chem. Commun., 2017, 53, 13264–13267 RSC.
- A. H. K. Al Temimi, R. van der Wekken-de Bruijne, G. Proietti, H. Guo, P. Qian and J. Mecinovic, Bioconjugate Chem., 2019, 30, 1798–1804 CrossRef CAS PubMed.
- A. H. K. Al Temimi, R. S. Teeuwen, V. Tran, A. J. Altunc, D. C. Lenstra, W. Ren, P. Qian, H. Guo and J. Mecinović, Org. Biomol. Chem., 2019, 17, 5693–5697 RSC.
- A. H. K. Al Temimi, H. I. V. Amatdjais-Groenen, Y. V. Reddy, R. H. Blaauw, H. Guo, P. Qian and J. Mecinović, Commun. Chem., 2019, 2, 112 CrossRef.
- J. C. Culhane, D. Wang, P. M. Yen and P. A. Cole, J. Am. Chem. Soc., 2010, 132, 3164–3176 CrossRef CAS PubMed.
- B. C. R. Dancy, S. A. Ming, R. Papazyan, C. A. Jelinek, A. Majumdar, Y. Sun, B. M. Dancy, W. J. Drury, R. J. Cotter, S. D. Taverna and P. A. Cole, J. Am. Chem. Soc., 2012, 134, 5138–5148 CrossRef CAS PubMed.
- T. Yang, X.-M. Li, X. Bao, Y. M. E. Fung and X. D. Li, Nat. Chem. Biol., 2016, 12, 70–72 CrossRef CAS PubMed.
- G. W. Langley, A. Brinko, M. Munzel, L. J. Walport, C. J. Schofield and R. J. Hopkinson, ACS Chem. Biol., 2016, 11, 755–762 CrossRef CAS PubMed.
- E. C. Gleeson, Z. J. Wang, S. D. Robinson, S. Chhabra, C. A. MacRaild, W. R. Jackson, R. S. Norton and A. J. Robinson, Chem. Commun., 2016, 52, 4446–4449 RSC.
- S. L. Mangold, D. J. O’Leary and R. H. Grubbs, J. Am. Chem. Soc., 2014, 136, 12469–12478 CrossRef CAS PubMed.
- G. de Bruin, E. J. van Rooden, D. Ward, C. Wesseling, A. M. C. H. van den Nieuwendijk, C. A. A. van Boeckel, C. Driessen, A. F. Kisselev, B. I. Florea, M. van der Stelt and H. S. Overkleeft, Eur. J. Org. Chem., 2017, 5921–5934 CrossRef CAS.
- E. J. Corey, F. Xu and M. C. Noe, J. Am. Chem. Soc., 1997, 119, 12414–12415 CrossRef CAS.
- F.-X. Theillet, S. Liokatis, J. O. Jost, B. Bekei, H. M. Rose, A. Binolfi, D. Schwarzer and P. Selenko, J. Am. Chem. Soc., 2012, 134, 7616–7619 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and full characterization of all compounds, HPLC and MALDI analyses, supporting figures, enzyme assays, peptide synthesis, protein production, synthesis, NMR data. See DOI: 10.1039/c9cc09098c |
| This journal is © The Royal Society of Chemistry 2020 |