
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA catalyst-free bis(triflyl)ethylation/benzannulation reaction: rapid access to carbazole-based superacidic carbon acids from alkynols†‡
Irene
Martín-Mejías
a,
Cristina
Aragoncillo
 b,
Hikaru
Yanai
*c,
Shoki
Hoshikawa
c,
Yuuki
Fujimoto
c,
Takashi
Matsumoto
c and
Pedro
Almendros
*a
b,
Hikaru
Yanai
*c,
Shoki
Hoshikawa
c,
Yuuki
Fujimoto
c,
Takashi
Matsumoto
c and
Pedro
Almendros
*a
aInstituto de Química Orgánica General, IQOG-CSIC, Juan de la Cierva 3, 28006-Madrid, Spain. E-mail: palmendros@iqog.csic.es
bGrupo de Lactamas y Heterociclos Bioactivos, Departamento de Química Orgánica, Unidad Asociada al CSIC, Facultad de Química, Universidad Complutense de Madrid, 28040-Madrid, Spain
cSchool of Pharmacy, Tokyo University of Pharmacy and Life Sciences, 1432-1 Horinouchi, Hachioji, Tokyo 192-0392, Japan. E-mail: yanai@toyaku.ac.jp
First published on 3rd January 2020
Abstract
Carbazoles possessing Tf2CHCH2 groups were obtained by the reaction of 1-(indol-2-yl)but-3-yn-1-ols with in situ-generated Tf2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 through vicinal difunctionalisation of the alkyne moiety, where the vinyl-type carbocation intermediate was selectively attacked by the indole moiety and not by the carbanion moiety.
CH2 through vicinal difunctionalisation of the alkyne moiety, where the vinyl-type carbocation intermediate was selectively attacked by the indole moiety and not by the carbanion moiety.
Carbanions are inherently reactive chemical species and the isolation of carbanion-containing salts is significantly limited. Delocalisation of the negative charge by an electron-withdrawing mesomeric (resonance) effect is a general approach for the improvement of thermodynamic stability. Some ‘free’ triphenylmethanides1 and cyclopentadienides2 have been isolated and characterised. Stabilisation of the carbanion is also achieved by substitution of the anionic carbon atom by fluorine atom(s) or fluorine-containing substituents. For example, Farnham and co-workers isolated and characterised a ‘free’ perfluorocarbanion.3 Carbanions bearing two triflyl groups (Tf = CF3SO2), which are known to be one of the strongest electron-withdrawing groups, attract much attention.4 Yanai reported several intramolecular salts 1–4 (Fig. 1A).5–9 For the stability of such [Tf2CR]− species, an orbital interaction between an occupied p orbital of the anionic carbon atom and adjacent σS–C(F3)* orbitals, called negative hyperconjugation,10 is pointed out to be a key factor (Fig. 1B).8 It is easily predictable that nucleophilicity of [Tf2CR]− is strongly suppressed due to the steric crowding around the anionic carbon atom and the charge-delocalised nature. Actually, compound 3 was the first example of a well-defined phosphorous carba-betaine, which did not undergo ring-closure into the corresponding phosphacyclopropane.8 Recently, a chemically inert property of [Tf2CR]− was applied to push–pull π systems. Push–pull ethylene 4 with a large distortion around the central C–C bond axis showed clearly charge-separated characters on the C–C bond in a complementary bonding analysis.9 These examples reveal that σ- or π-bonding with the [Tf2CR]− moiety is not favourable.
 | ||
| Fig. 1 (A) Zwitterions bearing a [Tf2CR]− structure, and (B) negative hyperconjugation in [Tf2CR]−. | ||
With such background, we were interested in the ring-closing reaction of vinyl-type carbocation INT-1 bearing a [Tf2CR]− moiety as the counter anion and a nucleophilic indole moiety (eqn (1)). As a pioneering work, Alcaide and Almendros reported a gem-bis(triflyl)cyclobutene synthesis through the stepwise (2+2) cycloaddition of several alkynes with Tf2CCH2,11,12 which was in situ-generated from 2-fluoropyridinium salt 2a7 developed by Yanai (eqn (2)).13 In the case of INT-1 derived from 1-(indol-2-yl)but-3-yn-1-ols 5 with Tf2CCH2, two reaction paths would be possible as follows: path (a) cyclobutene formation through a C–C bond forming reaction between the anionic carbon atom and the cationic C4 atom; path (b) carbazole formation induced by nucleophilic attack of the indole moiety on the C4 atom. Such a reaction system would allow further understanding of the chemical behaviour of the [Tf2CR]− species. In this paper, we report that the latter path selectively proceeds to give Tf2CHCH2-decorated carbazoles as the final products. Carbazoles are an important class of natural products, biologically active molecules, and advanced materials.14 Therefore, synthetic interests in them are renewed. In particular, transition metal-catalysed cyclisations of indolyl-3-alkyn-1-ols 5 have been developed to complement classical approaches (eqn (3)).15 The present results clearly demonstrate that non-ionic Tf2CCH2 effectively activates the internal alkynes in an electrophilic fashion. In addition, this is a rare example where attack of the non-ionic carbon-centred nucleophile is faster than that of the carbanion.
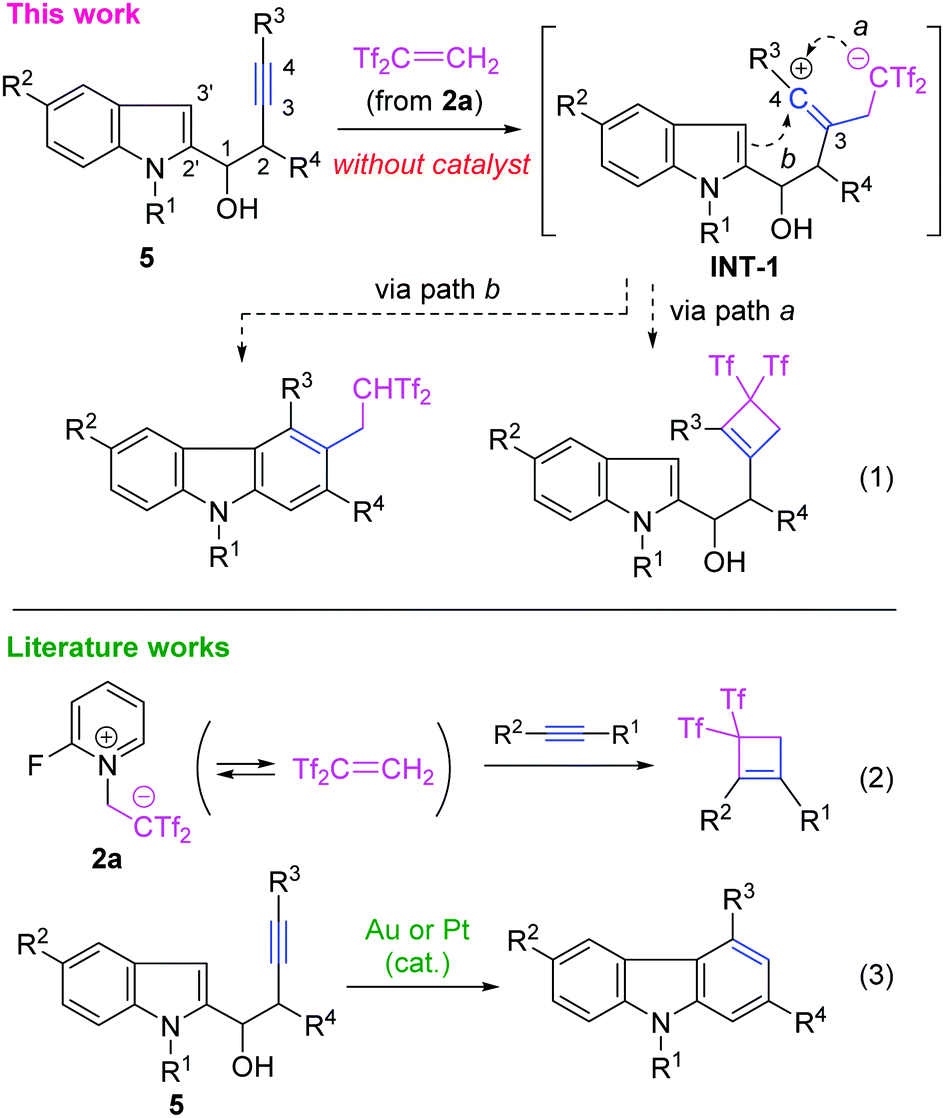
We first examined the reaction of indolyl-3-alkyn-1-ols 5 with 2-(2-fluoropyridinium-1-yl)-1,1-bis(triflyl)ethan-1-ide 2a under the optimised conditions for the cyclobutene-forming reaction (at 25 °C, in CH3CN)11 (Scheme 1). The reaction of (p-methoxyphenyl)alkyne 5a with 1.0 equiv. of 2a selectively produced the desired carbazole acid 6a-H, which was isolated as the corresponding sodium salt 6a in 56% yield after column chromatography on silica gel.16 In this case, the expected by-products including 3-bis(triflyl)ethylated indole and cyclobutene products were not obtained. For the present molecular transformation, the reaction solvents are critical. The use of DMF led to complexation of the reaction, while the reaction using ethanol or dichloromethane was not productive due to low solubility of 2a. During screening with several pyridinium salts 2b–e, we refound that a non-fluorinated salt 2e17 did not react with 5a under similar conditions and other 2-substituted pyridinium salts caused very slow conversion of 5a.7b Unfortunately, in the case of terminal alkyne 5b, 2,2-bis(triflyl)ethylation of the indole 3-position (C3′ atom) selectively occurred to give indole acid 7b-H, which was isolated as the corresponding triethylammonium salt 7b after column chromatography on silica gel pre-treated by Et3N for neutralisation.16 A similar result was observed in the reaction of phenylalkyne 5c. These results imply that alkyne-selectivity observed in the reaction of 5a is enhanced by the electron-donating aryl group on the alkyne terminus. Indeed, by applying common Lewis acids such as ZnI2 and InCl3, we observed no evidence of the formation of carbazoles from 5c. The adduct 7c gave only a complex mixture by heating at 80 °C. On this basis, we conclude that the 3-alkylated products 7 are formed in an irreversible manner and the possible carbazoles 6 did not generate from them.
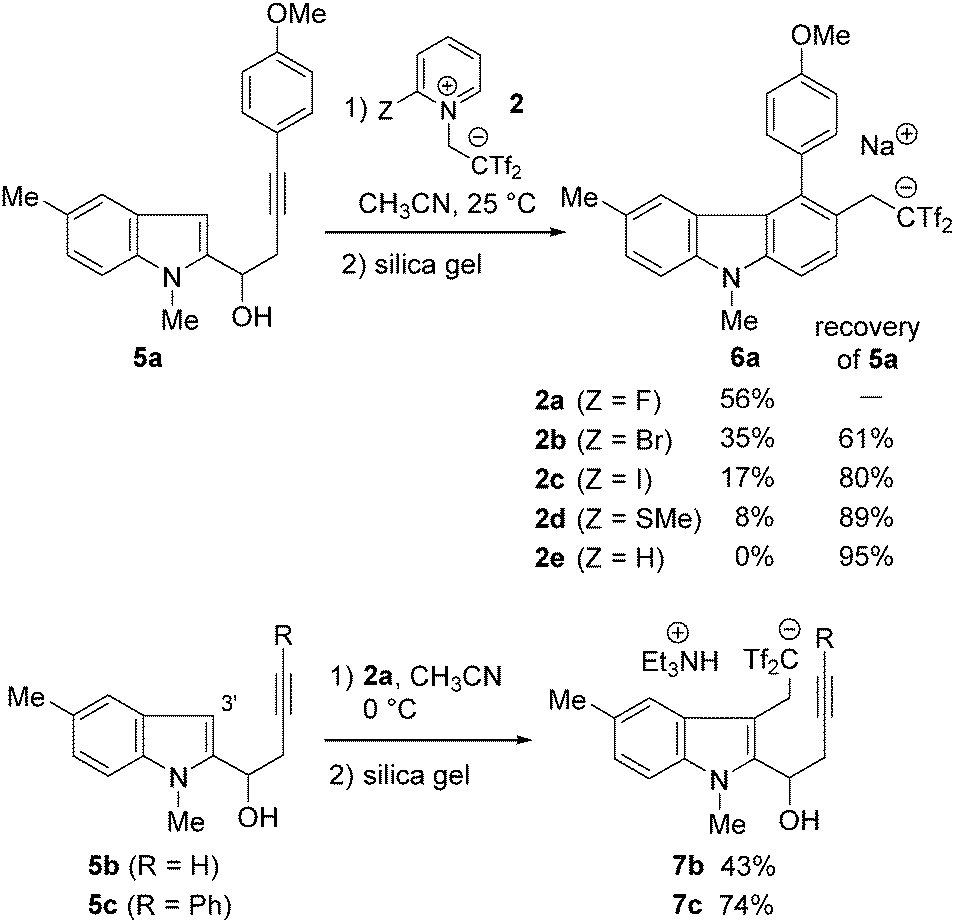 | ||
| Scheme 1 Reactions of indolyl-3-alkyn-1-ols 5a–c with 2a. | ||
The scope of the bis(triflyl)ethylation/benzannulation reaction was studied by using a series of indolyl-3-alkyn-1-ols 5 (Scheme 2). The reaction was applicable to 5-methylindoles 5d and 5e bearing electron-rich aryl groups including 2,4-dimethoxyphenyl and 2-thienyl groups on the alkyne terminus to give carbazoles 6d and 6e in moderate to good yields. 5-Unsubstituted indoles 5f–h and 5-methoxyindoles 5i and 5j were successfully converted to the desired carbazoles 6f–j. Likewise, 5-chloroindoles 5k and 5l reacted with Tf2CCH2 to give carbazoles 6k and 6l, respectively. Taking into account the low reactivity of the halogenated carbazoles in the direct aromatic electrophilic substitution (SEAr) reaction with in situ-generated Tf2CCH2 (vide infra), the present results show some synthetic advantages. The reactions of 2-methylbut-3-yn-1-ol 5m and N-Boc indole 5n also worked well to give carbazoles 6m and 6n, respectively. As shown in eqn (4), the reaction of iodoindole 5g-I with 2a gave 1-hydroxycarbazole 8. These examples suggest that the present reaction is potent for a regio-controlled synthesis of highly substituted carbazoles. The cyclisation methodology initiated by highly electrophilic Tf2CCH2 is not limited to carbazole formation. For example, the reaction of propargyl ether 9a with 2a produced 2H-chromene 10a in 95% yield (eqn (5)). Unfortunately, dialkyl alkyne 9b was significantly less reactive and its consumption was not observed even under heating conditions.

 | ||
| Scheme 2 Catalyst-free reaction of indolyl-3-alkyn-1-ols 5d–n with 2a. | ||
The Tf2CH-type superacidic molecules behave as carbon acids with unique catalytic activity.18,19 In the synthetic context,5 direct SEAr reaction of simple carbazoles with Tf2CCH2 would be attractive to obtain the carbazole-based acids.20 However, the reactions involving electron-deficient carbazoles 11a and 11b required heating conditions to form the products 12a and 12b (eqn (6); also, see ESI‡).
A plausible reaction pathway for carbazoles 6-H is shown in Scheme 3. First, the alkyne moiety of indolyl-3-alkyn-1-ols 5 traps Tf2CCH2 generated from reagent 2a to produce the zwitterionic vinyl-type carbocation INT-1. This initial addition is followed by a 5-endo-dig carbocyclisation through nucleophilic attack of the C2′ atom of the indole nucleus on the cationic C4 atom to give spirocyclic indolinium species INT-2.21 Next, a fused tricyclic intermediate INT-3 arises through 1,2-alkenyl migration in the spirocyclic nucleus of INT-2. Further aromatisation including deprotonation of INT-3 and dehydration of INT-4 should generate 6-H.
 | ||
| Scheme 3 Mechanistic explanation. | ||
This reaction pathway including 5-endo-dig cyclisation followed by ring-expansion is supported by a DFT simulation [PCM(CH3CN)-M06-2X/6-31+G(d) level of theory] of a model molecule 13, where the hydroxyl group of substrate 5g is replaced by a hydrogen atom (Fig. 2). For all optimised geometries, frequency calculations were conducted. The calculated Gibbs energy difference at 298 K (ΔG298) for the initial electrophilic attack of Tf2CCH2 on the alkynic C3 atom of 13 is only 16.3 kcal mol−1, when the initial state is used as a reference (Fig. 2). A vinyl-type carbocation INT-1′ corresponding to INT-1 shown in Scheme 3 was found as a local minimum species. The transition state geometry of the 5-endo-dig cyclisation (TS2) is very close to INT-1′, and the activation barrier to give INT-2′ is 1.9 kcal mol−1 (from INT-1′). In INT-1′, the C4–C2′ distance (275.1 pm) is obviously shorter than the C4–C3′ distance (320.7 pm). The low activation barrier and the geometric similarity between INT-1′ and TS2 support that the 5-endo-dig path rather than the 6-endo-dig path is kinetically favourable. In addition, the (2+2) cycloaddition path requires a much higher activation energy (6.3 kcal mol−1 from INT-1′; see, ESI‡). The following ring-expansion reaction (TS3) to give INT-3′ is highly exothermic and requires a small activation energy (7.0 kcal mol−1 from INT-2′). For each step on this pathway, the simulation well agrees with the fact that the reaction proceeds under really mild conditions.
 | ||
| Fig. 2 Reaction profile of the bis(triflyl)ethylation/benzannulation reaction. | ||
In conclusion, we successfully developed a sequential bis(triflyl)ethylation/benzannulation reaction to produce bis(triflyl)ethylated carbazoles from indolyl-3-alkyn-1-ols. This molecular transformation is triggered by a regioselective electrophilic attack of Tf2CCH2, which is generated from the 2-fluoropyridinium salt 2a in an in situ manner, on the alkyne moiety of the substrates. In vinyl-type carbocations thus generated, a ring-closing reaction between the cationic carbon atom and indole C2′ atom predominantly takes place. This event clearly shows the chemically inert properties of [Tf2CR]− in the chemical reaction.
Financial support for this work by AEI (MICIU) and FEDER (Project PGC2018-095025-B-I00) and KAKENHI (17K08224) is gratefully acknowledged. I. M. thanks MINECO for a predoctoral contract. We are grateful to M. Moyano for preliminary studies, Dr M. P. Ruiz and Dr C. Lázaro-Milla for helpful discussions, and Prof. B. Alcaide for continued support.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- (a) W. Schlenk and J. Holtz, Ber. Dtsch. Chem. Ges., 1916, 49, 603 CrossRef CAS; (b) S. Harder, Chem. – Eur. J., 2002, 8, 3229 CrossRef CAS; (c) M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1985, 107, 2174 CrossRef CAS.
- (a) E. Le Goff and R. B. LaCount, J. Am. Chem. Soc., 1963, 85, 1354 CrossRef CAS; (b) S. Matsumura and S. Seto, Chem. Pharm. Bull., 1963, 11, 126 CrossRef CAS PubMed; (c) R. Kuhn and D. Rewicki, Angew. Chem., 1967, 79, 648 CrossRef; (d) K. Okamoto, T. Kitagawa, K. Takeuchi, K. Komatsu and K. Takahashi, J. Chem. Soc., Chem. Commun., 1985, 173 RSC; (e) C. D. Gheewala, M. A. Radtke, J. Hui, A. B. Hon and T. H. Lambert, Org. Lett., 2017, 19, 4227 CrossRef CAS.
- (a) W. B. Farnham, Chem. Rev., 1996, 96, 1633 CrossRef CAS PubMed; (b) W. B. Farnham, D. A. Dixon and J. C. Calabrese, J. Am. Chem. Soc., 1988, 110, 2607 CrossRef CAS.
- (a) A. Hasegawa, T. Ishikawa, K. Ishihara and H. Yamamoto, Bull. Chem. Soc. Jpn., 2005, 78, 1401 CrossRef CAS; (b) D. Höfler, R. Goddard, J. B. Lingnau, N. Nöthling and B. List, Angew. Chem., Int. Ed., 2018, 57, 8326 CrossRef; (c) L. Turowsky and K. Seppelt, Inorg. Chem., 1988, 27, 2135 CrossRef CAS.
- For a review, see: H. Yanai and T. Taguchi, J. Fluorine Chem., 2015, 174, 108 CrossRef CAS.
- H. Yanai, T. Yoshino, M. Fujita, H. Fukaya, A. Kotani, F. Kusu and T. Taguchi, Angew. Chem., Int. Ed., 2013, 52, 1560 CrossRef CAS PubMed.
- (a) H. Yanai, Y. Takahashi, H. Fukaya, Y. Dobashi and T. Matsumoto, Chem. Commun., 2013, 49, 10091 RSC; (b) H. Yanai, R. Takahashi, Y. Takahashi, A. Kotani, H. Hakamata and T. Matsumoto, Chem. – Eur. J., 2017, 23, 8203 CrossRef CAS.
- H. Yanai, P. Almendros, S. Takahashi, C. Lázaro-Milla, B. Alcaide and T. Matsumoto, Chem. – Asian J., 2018, 13, 1956 CrossRef CAS PubMed.
- H. Yanai, T. Suzuki, F. Kleemiss, H. Fukaya, Y. Dobashi, L. A. Malaspina, S. Grabowsky and T. Matsumoto, Angew. Chem., Int. Ed., 2019, 58, 8839 CrossRef CAS PubMed.
- (a) J. D. Roberts, R. L. Webb and E. A. McElhill, J. Am. Chem. Soc., 1950, 72, 408 CrossRef CAS; (b) O. Exner and S. Böhm, New J. Chem., 2008, 32, 1449 RSC; (c) G. Raabe, H.-J. Gais and J. Fleischhauer, J. Am. Chem. Soc., 1996, 118, 4622 CrossRef CAS.
- (a) B. Alcaide, P. Almendros, I. Fernández and C. Lázaro-Milla, Chem. Commun., 2015, 51, 3395 RSC; (b) B. Alcaide, P. Almendros and C. Lázaro-Milla, Chem. – Eur. J., 2016, 22, 8998 CrossRef CAS PubMed; (c) B. Alcaide, P. Almendros and C. Lázaro-Milla, Adv. Synth. Catal., 2017, 359, 2630 CrossRef CAS.
- B. Alcaide, P. Almendros and C. Lázaro-Milla, Chem. Commun., 2015, 51, 6992 RSC.
- (a) H. Yanai, M. Fujita and T. Taguchi, Chem. Commun., 2011, 47, 7245 RSC; (b) H. Yanai, H. Ogura, H. Fukaya, A. Kotani, F. Kusu and T. Taguchi, Chem. – Eur. J., 2011, 17, 11747 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. Li, Chem. – Eur. J., 2019, 25, 1142 CrossRef CAS PubMed; (b) X. Zhang and S. Ma, Isr. J. Chem., 2018, 58, 608 CrossRef CAS; (c) G. Sathiyan, E. K. T. Sivakumar, R. Ganesamoorthy, R. Thangamuthu and P. Sakthivel, Tetrahedron Lett., 2016, 57, 243 CrossRef CAS; (d) A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 112, 3193 CrossRef CAS PubMed; (e) J. Roy, A. K. Jana and D. Mal, Tetrahedron, 2012, 68, 6099 CrossRef CAS; (f) H. J. Jiang, J. Sun and J. L. Zhang, Curr. Org. Chem., 2012, 16, 2014 CrossRef CAS; (g) J. Li and A. G. Grimsdale, Chem. Soc. Rev., 2010, 39, 2399 RSC; (h) T. Janosik, N. Wahlstrom and J. Bergman, Tetrahedron, 2008, 64, 9159 CrossRef CAS; (i) H.-J. Knölker and K. R. Reddy, Chemistry and Biology of Carbazole Alkaloids, in The Alkaloids, ed. G. A. Cordell, Academic Press, Amsterdam, 2008, vol. 65, pp. 1–430 Search PubMed.
- (a) Y. Qiu, W. Kong, C. Fu and S. Ma, Org. Lett., 2012, 14, 6198 CrossRef CAS PubMed; (b) A. S. K. Hashmi, W. Yang and F. Rominger, Chem. – Eur. J., 2012, 18, 6576 CrossRef CAS PubMed; (c) Y. Qiu, J. Zhou, C. Fu and S. Ma, Chem. – Eur. J., 2014, 20, 14589 CrossRef CAS PubMed; (d) J. Zhou, Y. Qiu, J. Li, C. Fu, X. Zhang and S. Ma, Chem. Commun., 2017, 53, 4722 RSC.
- The most carbon acids were purified by column chromatography on acidic or Et3N-pretreated silica gel. Therefore, chemical yields refer to the weight of isolated salts (see, ESI‡).
- L. L. Barber Jr and R. J. Koshar, US Pat., 3962342, 1976 Search PubMed.
- (a) T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277 CrossRef CAS PubMed; (b) C. H. Cheon and H. Yamamoto, Chem. Commun., 2011, 47, 3043 RSC.
- For selected examples, see: (a) K. Ishihara, A. Hasegawa and H. Yamamoto, Angew. Chem., Int. Ed., 2001, 40, 4077 CrossRef CAS; (b) A. Hasegawa, Y. Naganawa, M. Fushimi, K. Ishihara and H. Yamamoto, Org. Lett., 2006, 8, 3175 CrossRef CAS PubMed; (c) A. Takahashi, H. Yanai and T. Taguchi, Chem. Commun., 2008, 2385 RSC; (d) H. Yanai, O. Kobayashi, K. Takada, T. Isono, T. Satoh and T. Matsumoto, Chem. Commun., 2016, 52, 3280 RSC; (e) T. Gatzenmeier, M. van Gemmeren, Y. Xie, D. Höfler, M. Leutzsch and B. List, Science, 2016, 351, 949 CrossRef CAS PubMed; (f) D. Höfler, M. van Gemmeren, P. Wedemann, K. Kaupmees, I. Leito, M. Leutzsch, J. B. Lingnau and B. List, Angew. Chem., Int. Ed., 2017, 56, 1411 CrossRef PubMed.
- P. Almendros, H. Yanai, S. Hoshikawa, C. Aragoncillo, C. Lázaro-Milla, M. Toledano-Pinedo, T. Matsumoto and B. Alcaide, Org. Chem. Front., 2018, 5, 3163 RSC.
- A related spirocyclic intermediate has been recently proposed on the gold-catalysed synthesis of carbazoles: B. Alcaide, P. Almendros, C. Aragoncillo, E. Busto, C. G. López-Calixto, M. Liras, V. A. de la Peña O’Shea, A. García-Sánchez and H. V. Stone, Chem. – Eur. J., 2018, 24, 7620 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Benito Alcaide on the occasion of his retirement. |
| ‡ Electronic supplementary information (ESI) available: Computational details, experimental procedures, characterization data of new compounds, crystallographic details, and copies of NMR spectra for all new compounds. CCDC 1911534. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc08930f |
| This journal is © The Royal Society of Chemistry 2020 |