
Ratiometric detection of amyloid-β aggregation by a dual-emissive tris-heteroleptic ruthenium complex†

Abstract
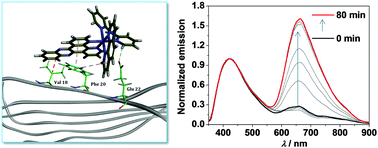
A dual-emissive tris-heteroleptic ruthenium complex is designed, synthesized and applied for the ratiometric photoluminescent detection of amyloid-β (Aβ) aggregation in both steady and transient states. The Aβ aggregation is supported by transmission electron microscopy and confocal laser scanning microscopy analysis. In addition, molecular docking calculations have been performed to gain insights into the interaction mode between the ruthenium complex and Aβ fibrils.
 Please wait while we load your content...
Please wait while we load your content...

