
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmorphous-to-crystalline transition and photoluminescence switching in guest-absorbing metal–organic network thin films†
Amr
Ghazy
 a,
Muhammad
Safdar
a,
Mika
Lastusaari
b and
Maarit
Karppinen
*a
a,
Muhammad
Safdar
a,
Mika
Lastusaari
b and
Maarit
Karppinen
*a
aDepartment of Chemistry and Materials Science, Aalto University, FI-00076 Espoo, Finland. E-mail: maarit.karppinen@aalto.fi
bDepartment of Chemistry, University of Turku, FI-20014 Turku, Finland
First published on 27th November 2019
Abstract
An amorphous metal–organic framework (aMOF) is an oxymoron as the porosity derived from the ordered network of the metal and organic moieties is the main characteristic of conventional crystalline MOFs. However, amorphous metal–organic materials can be synthesized from gaseous precursors through atomic/molecular layer deposition (ALD/MLD). We demonstrate an exciting interplay between luminescence properties and amorphous-to-crystalline transition realized upon water absorption in ALD/MLD aMOF films.
The rapidly expanding family of crystalline metal–organic framework (MOF) materials has been extensively investigated for several decades as exciting candidates for a breadth of applications, such as sensing, storage, catalysis, biomedicine and electronics.1,2 These materials are made of metal-ion nodes and organic linker molecules, and are characteristically highly porous; the manner in which the metal and organic constituent species are organized into the regular crystal lattice defines the important pore size distribution within the network. From this background, the concept of the “amorphous MOF (aMOF)” is self-contradicting; nevertheless, such materials have been gaining increasing interest in the recent years.3–10 An aMOF material has building units and connectivity modes similar to its crystalline counterpart (and thus shares similar chemistry) but lacks the long-range periodic order.
A great majority of crystalline MOFs are synthesized in the bulk (powder or single-crystal) form through solution-based routes; however, these conventional synthesis methods may not necessarily be ideal for the direct synthesis of aMOFs. The growth of crystalline MOFs from gaseous precursors has been demonstrated too; these techniques are particularly useful for the fabrication of high-quality thin films demanded to make the MOF materials technologically compatible with microelectronics.11–15 Even though not intentionally utilized so far, the gas-phase deposition of metal–organic thin films on suitable substrate surfaces could provide us with an ideal route for the direct aMOF synthesis. In particular, the currently strongly emerging atomic/molecular layer deposition (ALD/MLD) technique, developed from the state-of-the-art industrial ALD (atomic layer deposition)16 technology of inorganic thin films, has already been expanded to cover an impressive variety of metal species and organic moieties.17,18 In ALD/MLD, mutually reactive gaseous/vaporized metal and organic precursor molecules are sequentially pulsed into the reactor chamber; this enables – through self-limiting gas-surface chemical reactions – the fabrication of metal–organic materials with atomic/molecular level accuracy. The thus grown hybrid thin films are moreover precisely thickness-controlled, conformal and uniform over large-area and complex substrate surfaces, like the inorganic thin films deposited using the parent ALD technology. The metal–organic materials grown by ALD/MLD have already proven their potential e.g. in thermoelectric devices, lithium-ion batteries and catalysis.19–24 The technique has also been used to deposit lanthanide-based hybrid thin films, which have demonstrated interesting photoluminescence (Eu)25,26 and photon-upconversion (Yb,Er)27,28 properties.
Herein, we utilize the features of the combined ALD/MLD technique to fabricate novel amorphous neodymium terephthalate (Nd–TP) thin-film materials, from Nd(thd)3 (thd = 2,2,6,6-tetramethyl-3,5-heptanedione) and TPA (terephthalic acid; benzene-1,4-dicarboxylic acid) precursors. Previously, a similar ALD/MLD process for lanthanum yielded in situ crystalline La–TP thin films.29 We moreover demonstrate the amorphous-crystalline transition for our Nd–TP films upon water absorption. Neodymium is an attractive metal component for these hybrid thin films, as intense luminescence emission in the near-infrared (NIR) region has been reported for conventional crystalline Nd3+-based MOFs.30 This is of exclusive interest in bioimaging, bioassays, catalysis, NIR laser systems, and photon-upconversion.30–34 Here, we show excitingly that our ALD/MLD-grown Nd–TP thin films show photoluminescence switching upon aMOF-to-crystalline transition achieved through water absorption.
We carried out all the depositions on Si substrates at 200 °C based on our experience from the La(thd)3 + TPA process;29 at this deposition temperature homogeneous Nd–TP films were reproducibly obtained. The process was optimized for the precursor pulse lengths; Fig. 1a summarizes the data for films deposited with 300 ALD/MLD cycles. Pulse lengths of 4 s and 7.5 s for Nd(thd)3 and TPA, respectively, were sufficient to achieve the ideal surface-saturated chemical reactions of the precursors, with a growth-per-cycle (GPC) rate of 3.2 Å per cycle (calculated from the film thickness value obtained from X-ray reflectivity (XRR) analysis). For the rest of the experiments, we fixed the precursor pulse lengths to these values and used the following precursor/purge pulsing sequence: 4 s Nd(thd)3 → 5 s N2 → 7.5 s TPA → 25 s N2. The thus optimized deposition process showed the linear dependence of film thickness on the number of ALD/MLD cycles applied (Fig. 1b), which is one of the expected ALD/MLD trademarks.
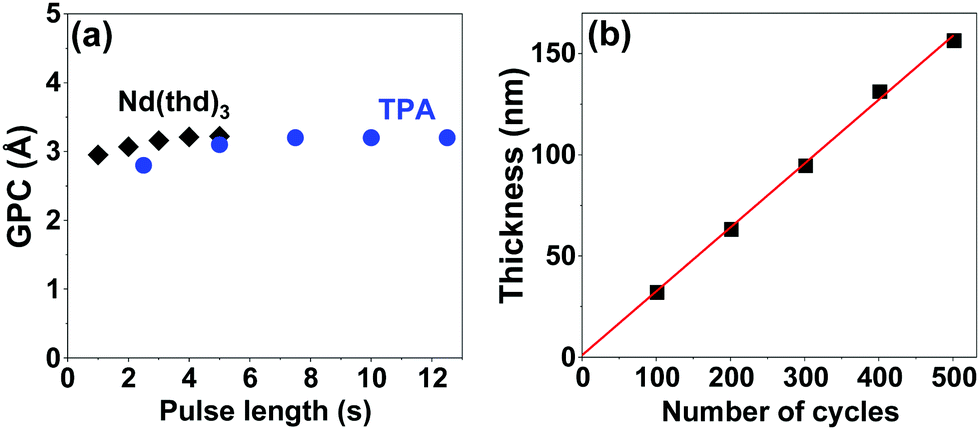 | ||
| Fig. 1 Optimization of the Nd(thd)3 + TPA process at 200 °C: (a) GPC as a function of the Nd(thd)3 and TPA pulse lengths, and (b) film thickness as a function of the number of ALD/MLD cycles of (4 s Nd(thd)3 → 5 s N2 → 7.5 s TPA → 25 s N2). | ||
For the further studies, we deposited the Nd–TP films with 300 ALD/MLD cycles; this resulted in ca. 95 nm thick films. The as-deposited films were perfectly amorphous based on grazing-incidence X-ray diffraction (GIXRD) observation, see Fig. 2a (the black pattern at the bottom). The Fourier transform infrared (FTIR) spectrum for the same film (Fig. 2b; black spectrum) confirmed the absence of free carboxylic acid groups (no peaks around 1700 cm−1) as expected as a consequence of the Nd–TP bond formation. Moreover the FTIR spectrum revealed the bridging-type bonding mode of the carboxylate groups (separation between the symmetric and asymmetric stretching vibrations: Δ = (1535–1385) cm−1 = 150 cm−1).35
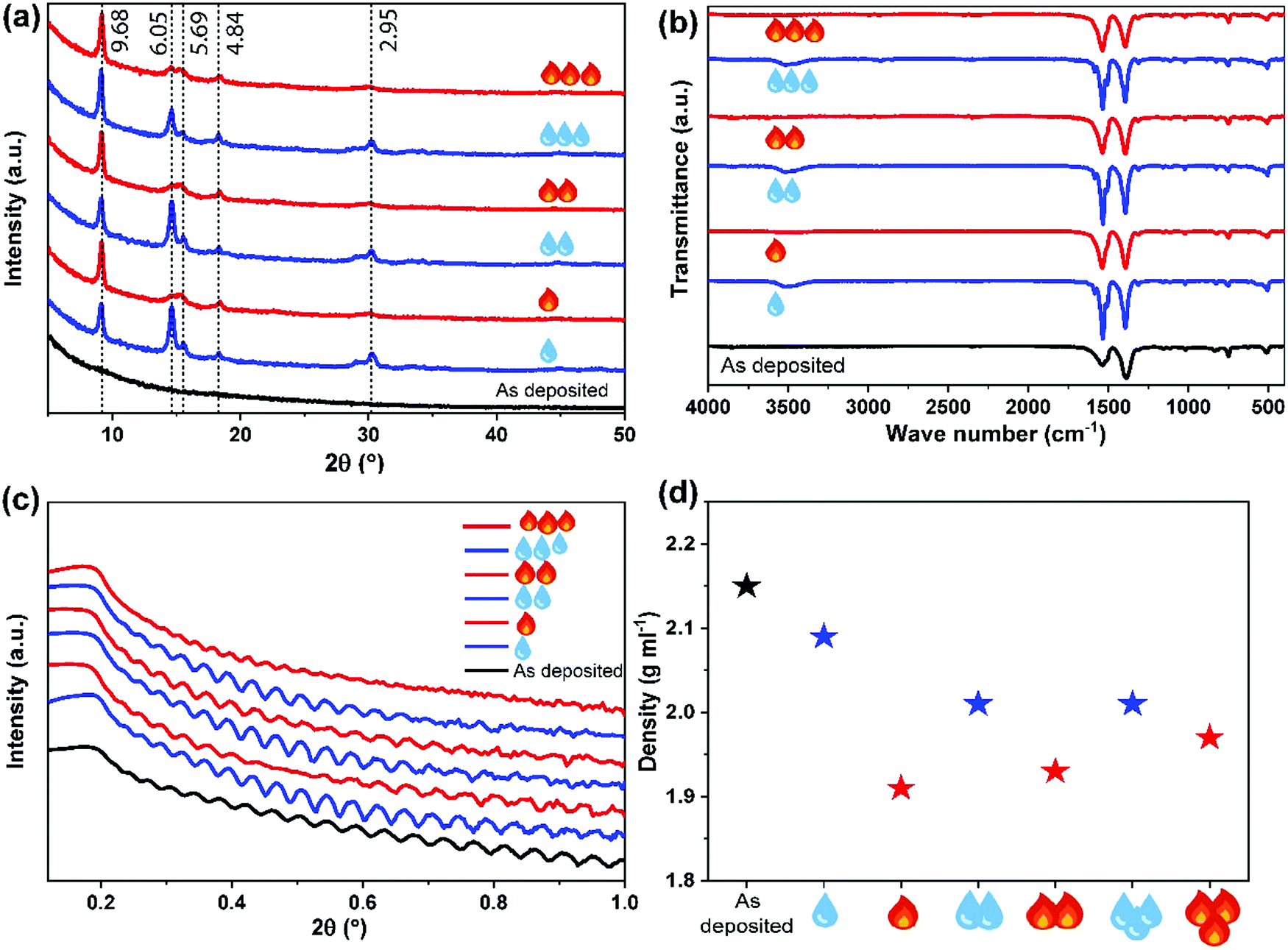 | ||
| Fig. 2 (a) GIXRD patterns along with the d-values of the main peaks, (b) FTIR spectra, (c) XRR curves, and (d) film density values determined from the XRR data for an as-deposited Nd–TP thin film (black), and for the same sample after three consecutive humidity (blue droplets) and heat (red flames) treatment cycles. | ||
We then exposed our as-deposited Nd–TP thin films to a room-temperature humidity treatment (RH 70%). Interestingly, this treatment caused the films to crystallize (Fig. 2a; blue pattern). FTIR data confirmed that upon the humidity treatment water is absorbed in the crystal lattice, see the wide peak appearing at around 3400 cm−1 in Fig. 2b;36 it should be noted that no indication of this peak was seen for the as-deposited film. The obtained GIXRD pattern showed close resemblance to the pattern calculated based on the crystal structure details reported for the terbium-based Tb2TP3(H2O)4 compound crystallizing in the triclinic (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) space group,37 see Fig. 3. In that structure carboxylate groups show bridging-type bonding mode, as was revealed for the present Nd–TP samples based on the FTIR data.
) space group,37 see Fig. 3. In that structure carboxylate groups show bridging-type bonding mode, as was revealed for the present Nd–TP samples based on the FTIR data.
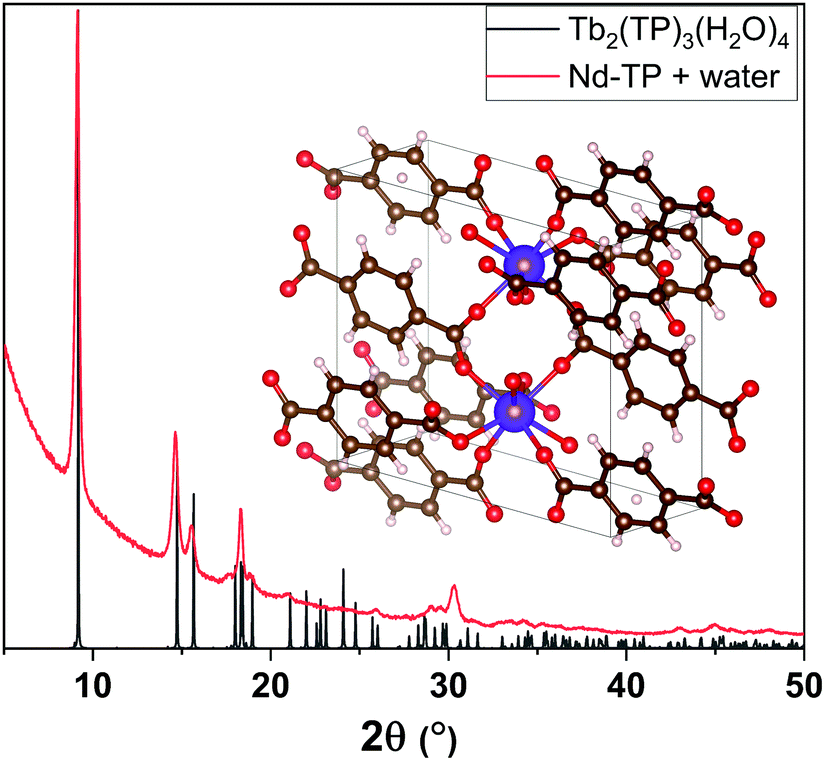 | ||
| Fig. 3 XRD peaks of water-containing Nd–TP thin films (red) match with the peaks from Tb2(TP)3(H2O)4 (grey) reported in the literature.37 | ||
To see whether we could remove the crystal water by heating without decomposing the hybrid material, we heat-treated the water-containing film overnight in air at 150 °C; indeed the ∼3400 cm−1 FTIR peak completely disappeared upon this heat treatment, while the film maintained its crystallinity. Moreover, we were able to demonstrate the reversibility of the water in-take/release behaviour by repeated humidity and heat treatments.
The humidity and heat treatments were repeated for the same sample three times, and the changes in crystallinity, composition, thickness and density were tracked using GIXRD, FTIR and XRR measurements, see Fig. 2. Upon these repeated treatments, the films remained crystalline, with a slight change only in the GIXRD pattern, in such a way that the low-angle peak (d = 9.68 Å) remained sharp and unchanged but the higher angle peaks were systematically sharper for the water-containing films and wider for the heat-treated ones. Similar systematic modifications could be revealed from the FTIR spectra as well, showing the broadening of the bands for the heat-treated films. Otherwise the main carboxylate symmetric and asymmetric stretching vibrations in the 1350–1550 cm−1 area remained essentially unaffected, i.e. the separation between these bands remained at 140–150 cm−1, confirming the bridging type mode of the carboxylate groups35 in all our Nd–TP films, both amorphous and crystalline (see the ESI† for details). It should be mentioned that Reineke et al.37 reported crystalline products with permanent microporosity after thermal liberation of water from their Tb2TP3(H2O)4 compound. No crystal structure details were given for the water-free Tb2TP3 structure; however, our GIXRD patterns for the heat-treated Nd–TP films are consistent with the XRD pattern reported for Tb2TP3.
The XRR patterns were used to determine the film thickness, density and roughness values of the samples after each humidity and heat treatment (see Fig. 2c, d and the ESI† for the fitting details). It could be seen that initially upon the first water absorption and consequent crystallization of the as-deposited aMOF Nd–TP film, the density decreased from 2.15 to 2.09 g cm−3; the surface roughness slightly increased (0.4 → 2.2 nm) as often seen when amorphous thin films are crystallized e.g. by thermal annealing. The decrease in density suggests that the crystal structure indeed is porous at least to some degree. Then, after the initial crystallization, the density systematically decreased when water was removed from the crystal structure and increased again upon water loading.
Finally, we investigated the photoluminescence characteristics (see the ESI† for measurement details) of our Nd–TP thin films. In particular, we wanted to see whether the aMOF-MOF transition and/or water intake would affect the emission intensity. First of all, the characteristic Nd3+ emission peaks at 880 nm (4F3/2 → 4I9/2) and 1064 nm (4F3/2 → 4I11/2) were seen for all the representative samples investigated (Fig. 4); note that the peak at 1064 nm is due to the Nd:YAG laser excitation source, except for the as-deposited film that seems to contribute to the emission in this spectral region as well (judging by the change in the peak shape). Most strikingly, the emission intensity at 880 nm is significantly higher for the as-deposited amorphous Nd–TP film compared to the two crystalline films. In the case of the water-containing (humidity-treated) film, this could be explained by non-radiative relaxation processes due to high-frequency vibrations of the –OH groups.38 However, since the removal of the crystalline water did not increase the luminescence intensity, but rather decreased it, it seems that the high-frequency vibrations of the –OH groups are not at least the main reason for the observed luminescence quenching. A more probable explanation is the decrease in film density upon the crystallization, which increases the inter-ionic distances and could thereby make the energy-transfer processes less efficient. Elucidation of the detailed mechanism definitely requires dedicated studies in future.
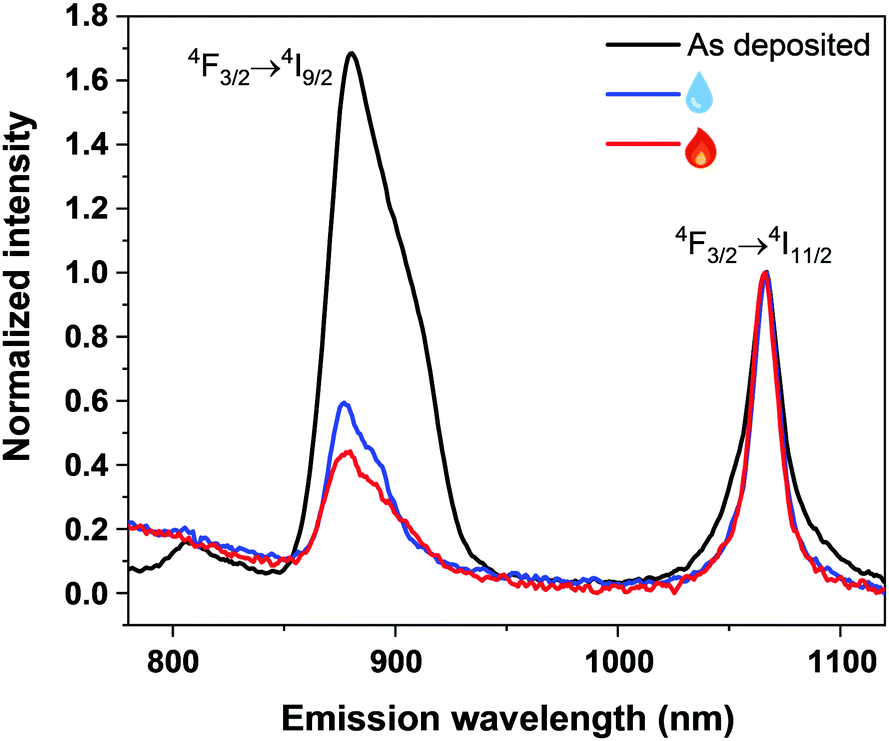 | ||
| Fig. 4 Photoluminescence emission spectra of the as-deposited (black), humidity-treated (blue) and subsequently heat-treated (red) Nd–TP thin films. | ||
In conclusion, we have demonstrated the deposition of amorphous neodymium terephthalate thin films through a single-step ALD/MLD process from gaseous (sublimated) Nd(thd)3 and TPA precursors at 200 °C. This process yields highly homogeneous Nd–TP thin films at a growth rate of ca. 3.2 Å per ALD/MLD cycle. The ALD/MLD method is thus believed to be uniquely advantageous for the fabrication of novel aMOF-type metal–organic materials with only short-range order. The as-deposited films then crystallized upon absorbing water when treated under humid conditions (70% RH), while a consequent heat treatment at 150 °C completely removed the coordinated water molecules but retained the film crystallinity. This reversible guest water molecule in-take and release could be repeated several times without losing the crystalline coordination network structure. This allowed us to investigate the possible effects of the aMOF-to-MOF type transition in our Nd–TP thin films. Most excitingly, the characteristic Nd3+ photoluminescence seen under laser excitation for the as-deposited amorphous films was found to be significantly reduced for the crystalline films. We believe that this type of switching action upon the guest-molecule induced amorphous-to-crystalline transition is an intriguing material functionality to be further elaborated towards novel applications.
We acknowledge the funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Advanced Grant Agreement (339478) and Academy of Finland (296299), and the use of the RawMatTERS Finland Infrastructure (RAMI) at Aalto University.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS.
- B. Li, H.-M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS PubMed.
- T. D. Bennett, A. L. Goodwin, M. T. Dove, D. A. Keen, M. G. Tucker, E. R. Barney, A. K. Soper, E. G. Bithell, J. C. Tan and A. K. Cheetham, Phys. Rev. Lett., 2010, 104, 115503 CrossRef PubMed.
- T. D. Bennett, P. Simoncic, S. A. Moggach, F. Gozzo, P. MacChi, D. A. Keen, J. C. Tan and A. K. Cheetham, Chem. Commun., 2011, 47, 7983–7985 RSC.
- K. Ohara, J. Martí-Rujas, T. Haneda, M. Kawano, D. Hashizume, F. Izumi and M. Fujita, J. Am. Chem. Soc., 2009, 131, 3860–3861 CrossRef CAS PubMed.
- C. Orellana-Tavra, E. F. Baxter, T. Tian, T. D. Bennett, N. K. H. Slater, A. K. Cheetham and D. Fairen-Jimenez, Chem. Commun., 2015, 51, 13878–13881 RSC.
- X. Zhang, H. Li, X. Lv, J. Xu, Y. Wang, C. He, N. Liu, Y. Yang and Y. Wang, Chem. – Eur. J., 2018, 24, 8822–8832 CrossRef CAS PubMed.
- W. Niu and Y. Yang, ACS Appl. Energy Mater., 2018, 1, 2440–2445 CrossRef CAS.
- T. D. Bennett and A. K. Cheetham, Acc. Chem. Res., 2014, 47, 1555–1562 CrossRef CAS PubMed.
- T. D. Bennett and S. Horike, Nat. Rev. Mater., 2018, 3, 431–440 CrossRef.
- E. Ahvenniemi and M. Karppinen, Chem. Commun., 2016, 52, 1139–1142 RSC.
- E. Ahvenniemi and M. Karppinen, Chem. Mater., 2016, 28, 6260–6265 CrossRef CAS.
- M. D. Allendorf, A. Schwartzberg, V. Stavila and A. A. Talin, Chem. – Eur. J., 2011, 17, 11372–11388 CrossRef CAS PubMed.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- A. Khayyami, A. Philip and M. Karppinen, Angew. Chem., Int. Ed., 2019, 58, 13400–13404 CrossRef CAS PubMed.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- K. B. Klepper, O. Nilsen and H. Fjellvåg, Dalton Trans., 2010, 39, 11628–11635 RSC.
- P. Sundberg and M. Karppinen, Beilstein J. Nanotechnol., 2014, 5, 1104–1136 CrossRef PubMed.
- T. Tynell, I. Terasaki, H. Yamauchi and M. Karppinen, J. Mater. Chem. A, 2013, 1, 13619–13624 RSC.
- T. Tynell, A. Giri, J. Gaskins, P. E. Hopkins, P. Mele, K. Miyazaki and M. Karppinen, J. Mater. Chem. A, 2014, 2, 12150–12152 RSC.
- M. Nisula and M. Karppinen, Nano Lett., 2016, 16, 1276–1281 CrossRef CAS PubMed.
- M. Nisula and M. Karppinen, J. Mater. Chem. A, 2018, 6, 7027–7033 RSC.
- J. Heiska, M. Nisula and M. Karppinen, J. Mater. Chem. A, 2019, 7, 18735–18758 RSC.
- C. MacIsaac, J. R. Schneider, R. G. Closser, T. R. Hellstern, D. S. Bergsman, J. Park, Y. Liu, R. Sinclair and S. F. Bent, Adv. Funct. Mater., 2018, 28, 1800852 CrossRef.
- Z. Giedraityte, P. Sundberg and M. Karppinen, J. Mater. Chem. C, 2015, 3, 12316–12321 RSC.
- Z. Giedraityte, J. Sainio, D. Hagen and M. Karppinen, J. Phys. Chem. C, 2017, 121, 17538–17545 CrossRef CAS.
- Z. Giedraityte, M. Tuomisto, M. Lastusaari and M. Karppinen, ACS Appl. Mater. Interfaces, 2018, 10, 8845–8852 CrossRef CAS PubMed.
- M. Tuomisto, Z. Giedraityte, L. Mai, A. Devi, V. Boiko, K. Grzeszkiewicz, D. Hreniak, M. Karppinen and M. Lastusaari, J. Lumin., 2019, 213, 310–315 CrossRef CAS.
- J. Penttinen, M. Nisula and M. Karppinen, Chem. – Eur. J., 2017, 23, 18225–18231 CrossRef CAS PubMed.
- L. N. Jia, L. Hou, L. Wei, X. J. Jing, B. Liu, Y. Y. Wang and Q. Z. Shi, Cryst. Growth Des., 2013, 13, 1570–1576 CrossRef CAS.
- M. Gustafsson, A. Bartoszewicz, B. Martiín-Matute, J. Sun, J. Grins, T. Zhao, Z. Li, G. Zhu and X. Zou, Chem. Mater., 2010, 22, 3316–3322 CrossRef CAS.
- S. V. Eliseeva and J. C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- D. F. Sava Gallis, L. E. S. Rohwer, M. A. Rodriguez, M. C. Barnhart-Dailey, K. S. Butler, T. S. Luk, J. A. Timlin and K. W. Chapman, ACS Appl. Mater. Interfaces, 2017, 9, 22268–22277 CrossRef CAS PubMed.
- H. Xu, R.-Z. Jin, C.-Y. Wu, Y. Yang and G.-D. Qian, Spectrosc. Spect. Anal., 2008, 28, 1734–1736 CAS.
- C. A. Téllez S, E. Hollauer, M. Mondragon and V. M. Castaño, Spectrochim. Acta, Part A, 2001, 57, 993–1007 CrossRef.
- K. B. Klepper, O. Nilsen, S. Francis and H. Fjellvåg, Dalton Trans., 2014, 43, 3492–3500 RSC.
- T. M. Reineke, M. Eddaoudi, M. Fehr, D. Kelley and O. M. Yaghi, J. Am. Chem. Soc., 1999, 121, 1651–1657 CrossRef CAS.
- T. Cong, Y. Ding, S. Xin, X. Hong, H. Zhang and Y. Liu, Langmuir, 2016, 32, 13200–13206 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc08904g |
| This journal is © The Royal Society of Chemistry 2020 |