
A highly efficient electrocatalyst based on double perovskite cobaltites with immense intrinsic catalytic activity for water oxidation†

Abstract
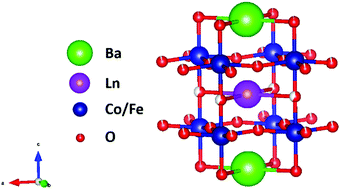
High temperature electrocatalysts based on double perovskite cobaltites that are typically employed in proton ceramic fuel cells and electrolyzers are exploited here for room temperature water oxidation. The double perovskites are assessed by the RctCdl product and we show that their intrinsic catalytic activities exceed that of IrO2.
 Please wait while we load your content...
Please wait while we load your content...

