
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,3-Carboboration of iodonium ylides†‡
Theodore A.
Gazis
 a,
Bayan A. J.
Mohajeri Thaker
a,
Darren
Willcox
ab,
Darren M. C.
Ould
a,
Jan
Wenz
a,
Jeremy M.
Rawson
c,
Michael S.
Hill
d,
Thomas
Wirth
e and
Rebecca L.
Melen
*a
a,
Bayan A. J.
Mohajeri Thaker
a,
Darren
Willcox
ab,
Darren M. C.
Ould
a,
Jan
Wenz
a,
Jeremy M.
Rawson
c,
Michael S.
Hill
d,
Thomas
Wirth
e and
Rebecca L.
Melen
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, Cymru/Wales CF10 3AT, UK. E-mail: MelenR@cardiff.ac.uk
bDepartment of Chemistry, University of Manchester, Oxford Rd, Manchester, M13 9PL, UK
cDepartment of Chemistry and Biochemistry, University of Windsor, 401 Sunset Avenue, Windsor, Ontario N9B 3P4, Canada
dDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
eSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, Cymru/Wales CF10 3AT, UK
First published on 14th February 2020
Abstract
Herein, we disclose the utilisation of iodonium ylides to access a range of boron dienolates. Heating of acyclic iodonium ylides in the presence of different aryl boranes leads to the formation of rare 1,3-carboboration products. This methodology could not be expanded to cyclic iodonium ylides which instead formed a Lewis acid–base adduct. Products proved to be remarkably stable under a wide range of conditions allowing for their long term storage.
The versatility of organoboranes in synthesis is beyond dispute.1 From reaction intermediates (amination reactions2) to substrates (cross-coupling reactions3) and catalysts (e.g. Lewis acid catalysis4 or frustrated Lewis pair chemistry5) they represent an invaluable tool in the arsenal of the synthetic chemist. One such class of boranes, which has come to the forefront of modern Lewis acid chemistry, is that of the highly electrophilic fluorophenyl boranes.6 These boranes demonstrate relatively high stability towards boron–carbon hydrolytic cleavage. However, the bond is sufficiently labile to allow for its utilisation in a range of carboborations of sp- and sp2-hybridised bonds.7 Of these, 1,1-carboboration is the most widely explored, partly thanks to the Wrackmeyer reaction, and enables facile access to substituted olefins.8
To date, alkynes remain the ‘go-to’ substrate for 1,1-carboborations with numerous examples reported.9 However, recently, organic azides have been shown to undergo 1,1-carboborations with B(C6F5)3 to generate imines. This reaction was shown to proceed via an initial adduct formation between B(C6F5)3 and RN3 followed by a Staudinger type reaction (Scheme 1a).10 Conversely, metal-free 1,2-carboboration reactions are much rarer, especially for sp-hybridised systems which to date are limited to highly electrophilic borocations.11 1,2-Carboboration of sp2-centres has been exemplified through the use of allenyl esters and ketones with B(C6F5)312 and recently isocyanates leading to 4-membered cyclic compounds exhibiting frustrated Lewis pair characteristics (Scheme 1b).13 1,2-Carboboration of cyclic iminium ions with B(C6F5)3, has also been reported under photochemical conditions leading to highly desirable tertiary amines.14 These transformations demonstrate the proclivity of B(C6F5)3 to effect selectively 1,2-aryl transfers. However, the corresponding 1,3-aryl transfers are still an under-represented class of transformations. Aryl transfer reactions utilising electrophilic boranes and diazo species have been reported by Stephan, however these reactions were limited to the transfer of the C6F5 moiety.15
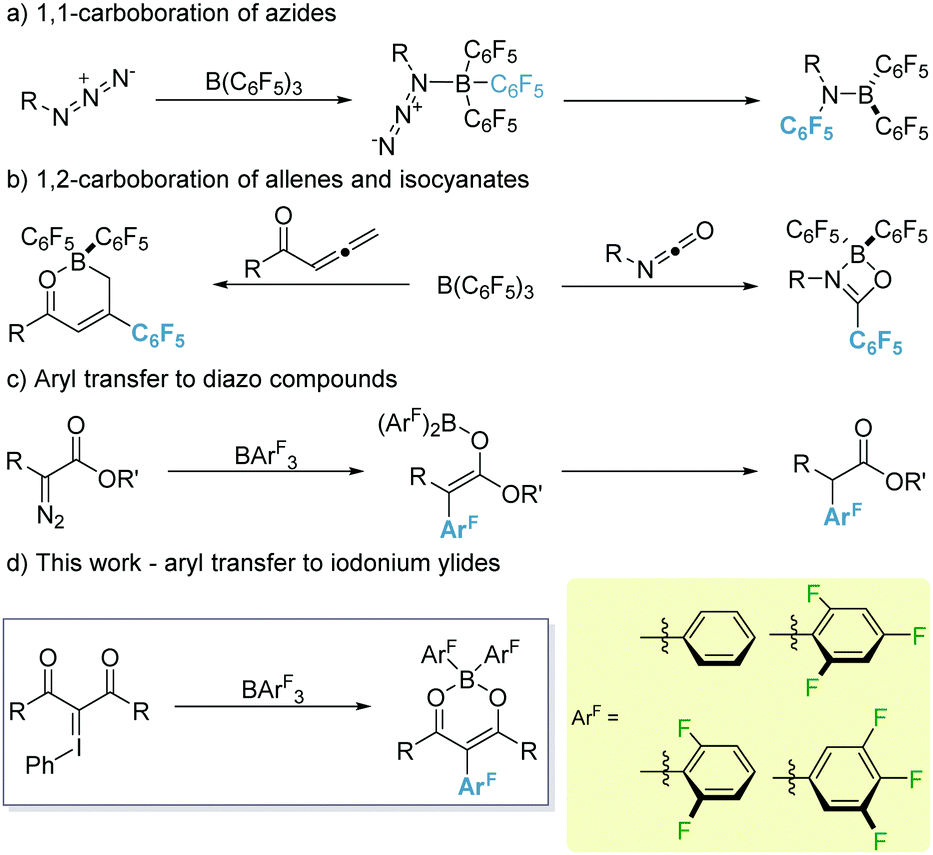 | ||
| Scheme 1 Reported aryl transfer reactions from electrophilic boranes (a–c) and this work (d). | ||
Recently, our group reported that transfer of other halogenated aryl groups to diazo species is possible (Scheme 1c).16 Given the analogous nature between diazo species and iodonium ylides as carbene precursors,17 we envisaged that a similar aryl transfer should be viable. To this end, we divulge the reaction of iodonium ylides with a library of boranes (BArF3; ArF = 3,4,5-F3C6H2 (1a), 2,4,6-F3C6H2 (1b), 2,6-F2C6H3 (1c), C6H5 (1d) and C6F5 (1e)) displaying varying levels of Lewis acidity (Scheme 1d).
Initial studies of the reaction of dimethyl 2-(phenyl-λ3-iodaneylidene)malonate [PhIC(CO2Me)2] 2a with borane 1a in refluxing dichloromethane led to a pale-yellow homogeneous solution. Subsequent workup and isolation afforded 3a as colourless crystals in 45% yield. The 11B{1H} NMR spectrum of 3a shows a broad resonance at δ = 7.5 ppm, while the 1H and 19F NMR spectra were consistent with the incorporation of the 3,4,5-trifluorophenyl moiety showing two C6H2F3 ring environments in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The yield could be improved by conducting the reaction in either chloroform or toluene (56% and 74% respectively) at 50 °C. Recrystallisation from a dichloromethane/pentane solution led to single crystals whose exact nature was elucidated by single crystal X-ray diffraction (Fig. 1). Compound 3a crystallised in the P
:1 ratio. The yield could be improved by conducting the reaction in either chloroform or toluene (56% and 74% respectively) at 50 °C. Recrystallisation from a dichloromethane/pentane solution led to single crystals whose exact nature was elucidated by single crystal X-ray diffraction (Fig. 1). Compound 3a crystallised in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with four molecules present in the unit cell. The solid-state structure shows the C2O2B ring is non-planar and instead adopts an envelope-type geometry, with the boron heteroatom sitting out of the plane and a fold angle (the angle between the OBO and OCCO mean planes) of 23.3(3)°. The boron heteroatom is four coordinate and is slightly distorted from a tetrahedral geometry, with an O–B–O bond angle of 107.52(18)° (see ESI‡ for further bond angles). The B–O bond lengths (1.523(3) Å and 1.525(3) Å) were found to be slightly longer than typically observed (1.468 Å in BO4−)18 and the C–O bond lengths were found to be 1.287(3) and 1.288(3) Å, which is approximately intermediate between a single and double C–O bond character.18 This suggests resonance character is exhibited in the core C2O2B six-membered ring, which is further observed in the C–C bond lengths, which are again in between single and double bond character (1.389(3) Å and 1.396(3) Å). These metrics are comparable to related diketone structures reported by Chujo.19
space group with four molecules present in the unit cell. The solid-state structure shows the C2O2B ring is non-planar and instead adopts an envelope-type geometry, with the boron heteroatom sitting out of the plane and a fold angle (the angle between the OBO and OCCO mean planes) of 23.3(3)°. The boron heteroatom is four coordinate and is slightly distorted from a tetrahedral geometry, with an O–B–O bond angle of 107.52(18)° (see ESI‡ for further bond angles). The B–O bond lengths (1.523(3) Å and 1.525(3) Å) were found to be slightly longer than typically observed (1.468 Å in BO4−)18 and the C–O bond lengths were found to be 1.287(3) and 1.288(3) Å, which is approximately intermediate between a single and double C–O bond character.18 This suggests resonance character is exhibited in the core C2O2B six-membered ring, which is further observed in the C–C bond lengths, which are again in between single and double bond character (1.389(3) Å and 1.396(3) Å). These metrics are comparable to related diketone structures reported by Chujo.19
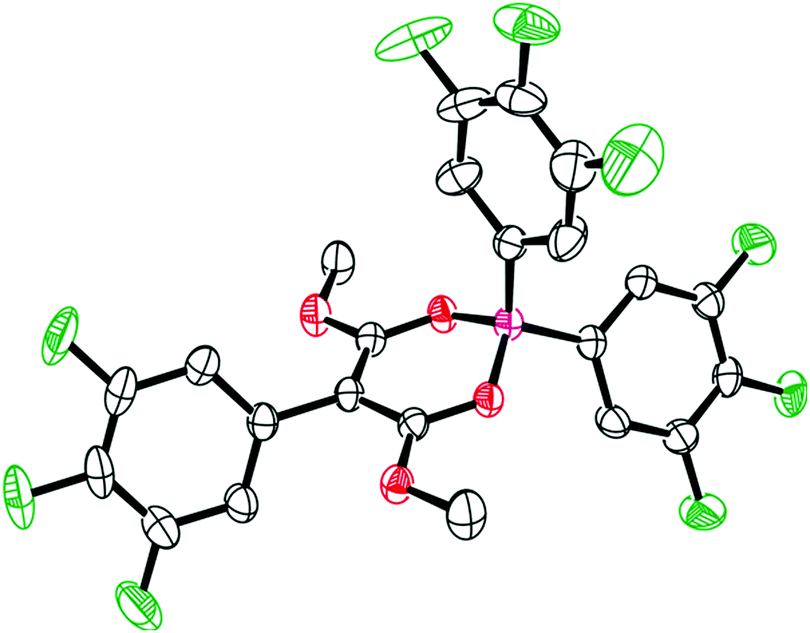 | ||
| Fig. 1 Solid-state structure of 3a. C: black, B: pink, O: red, F: green. Thermal ellipsoids drawn at 50% probability. H atoms omitted for clarity. | ||
The analogous reaction of hypervalent iodine starting material 2a with equimolar quantities of boranes 1b–d proceeded in a similar fashion furnishing the aryl transfer products in moderate to good yields (up to 67%) (Scheme 2). All products (3a–d) exhibited a broad resonance in the 11B{1H} NMR spectrum between δ = 6.6 and 9.2 ppm. As with 3a, 1H NMR data confirmed the incorporation of the fluoraryl moiety and the 19F spectrum resonances were consistent with two inequivalent fluoroaryl rings. Where yields are concerned, a tentative relationship to Lewis acidity could be made. As shown in Scheme 2, an increase in borane Lewis acidity (as determined by the Gutmann–Beckett method)20 provided a seemingly positive effect on the product yield. A notable exception is the reaction with B(C6F5)3 (1e) which led to an inseparable mixture of products (see ESI‡ for crystal data of 3e), despite having a Lewis acidity comparable to B(3,4,5-C6F3H2)3. The characterisation of by-products and intermediates is always useful in evaluating mechanistic pathways. In this context we re-examined the reaction of both 1b and 1e with 2a on a larger scale as these produced the lowest isolated yields. Multinuclear NMR studies provided complex spectra from which no obvious hydrolysis product or reaction intermediate could be unambiguously identified. Attempted recrystallisations using layering and solvent diffusion techniques from a variety of solvent mixtures also proved unsuccessful.
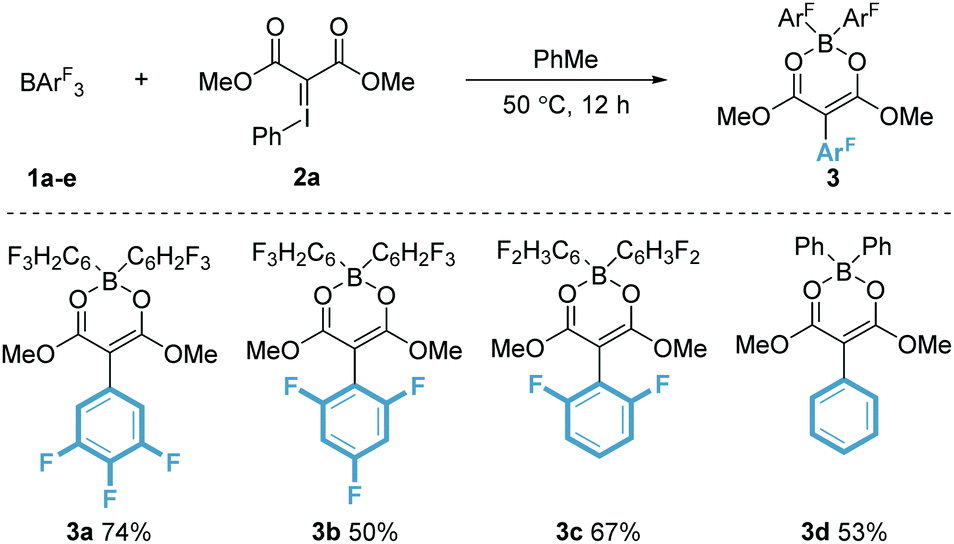 | ||
| Scheme 2 Reaction of 2a with boranes 1a–d. | ||
Having established the feasibility of transferring fluorophenyl rings to symmetrical bis-ester stabilised ylides, we turned our attention to less stable iodonium ylides bearing an ester and a ketone (2b), as well as a diketone (2c) (Scheme 3). As before, reaction of 2b and 2c with tris(3,4,5-trifluorophenyl)borane (1a) showed similar reactivity to that with 2a resulting in the formation of 3f and 3g respectively. The 11B{1H} NMR spectrum showed a broad resonance at δ = 7.7 ppm for 3f and δ = 7.3 ppm for 3g consistent with a four-coordinate boron centre with diminished Lewis acidity.
 | ||
| Scheme 3 Reaction of iodonium ylides 2b and 2c with B(3,4,5-F3C6H2)31a. | ||
1H and 19F NMR spectroscopic data were also consistent with the formation of 3f and 3g. X-ray crystallography unambiguously confirmed the structures of both products (Fig. 2). Compound 3f crystallised in the P21/c space group and has similar metrics to that of 3a, with a distorted tetrahedral four-coordinate boron and B–O bond lengths 1.488(3) Å and 1.540(3) Å. It was noted that the degree of resonance appeared less in 3g than 3a, with C–O bond lengths of 1.278(3) Å and 1.316(3) Å and C–C bond lengths of 1.366(4) Å and 1.419(4) Å. Compound 3g, like 3a, crystallised in the P space group and again has similar metrics to 3a (see ESI‡ for further details). Repeated attempts were made to cleave the boron moiety from compounds 3a–g to yield organic products in which an aryl functionality had been incorporated. However, these compounds proved remarkably stable and resistant to hydrolysis in both acidic and aqueous media,21e.g. deprotection using a variety of reagents all proved unsuccessful including: (i) NaOH/H2O2; (ii) Ag2O/BnBr in MeCN; (iii) N2H4·H2O/AcOH in EtOH (to form the equivalent pyrazole); (iv) Na2CO3 in EtOH, H2O then HCl. It was postulated that removing the ylide's ability to form stable intramolecular chelates would facilitate such a cleavage. To test this, iodonium ylide 2d bearing a dimedone motif was synthesised. Due to its conformational rigidity it was envisaged that only a single oxygen atom would be available to yield a B–O linkage thus facilitating hydrolysis. Yet, when 2d was reacted with all boranes 1, unlike the acyclic iodonium ylides, a complex mixture of products was formed in most cases. With B(C6F5)3 (1e) however adduct 4 was isolated and crystallised (see ESI‡). This adduct proved to be resistant to elevated temperatures (90 °C), precluding the carboboration reaction. This indicates that a single B–O connection is not sufficient to activate the boron and effect the transfer. However secondary effects should not be discounted. It is reported that cyclic iodonium ylides are more stable compared to their open chain counterparts. This effect is imparted by secondary C–I⋯O bonding.22 However, this is a weak interaction that should be readily disrupted at elevated temperatures. It is likely these two factors work in a synergistic fashion preventing the aryl transfer. Adduct 4 shows short intermolecular O⋯I contacts in the solid-state at 2.916 Å and 3.105 Å, slightly longer than those reported for similar compounds in the absence of the B(C6F5)3 adduct (Scheme 4).22
 | ||
| Fig. 2 Solid-state structure of 3f and 3g. C: black, B: pink, O: red, F: green. Thermal ellipsoids drawn at 50% probability. H atoms omitted for clarity. | ||
 | ||
| Scheme 4 Reaction of 2d with B(C6F5)31e. | ||
In order to probe the reactivity of the Lewis acidic borane with the iodonium ylide, a series of computational studies on the formation of 3c were undertaken with the M06-2X functional and LACVP+ basis set (see ESI‡ for details). Initial calculations probed adduct formation between the borane and the iodonium ylide using the carbonyl O atom as a hard donor. Adduct formation was computed to be strongly favourable (ΔE = −115 kJ mol−1) and an analysis of the two conformers revealed that the less sterically hindered conformation was more favoured by just 11 kJ mol−1 (Scheme 5). Reaction profiles for migration of the aryl group to the ylidic carbon commencing from both exo and endo adducts were explored and the migration commencing from the more stable exo adduct passed through a lower energy transition state. These studies reveal a four-coordinate transition state with limited C–I bond weakening (dC⋯I = 2.10 Å) reflecting an early transition state in agreement with Hammond's postulate.23 After elimination of the aryl iodide leaving group, cyclisation exhibits a low activation energy and leads to the observed ring-closed product as the most stable species on the potential energy surface.
 | ||
Scheme 5 Reaction profile for the formation of 3c from B(C6H3F2)3 and C6H3F2I![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(COOMe)2. C(COOMe)2. | ||
In summary, we have showcased an innovative route to boron dienolates utilising iodonium ylides. This study revealed that acyclic symmetrical iodonium ylides were successful substrates. Unsymmetrical ylides were also amenable to this transformation, whereas conformationally restricted cyclic variants led to adduct formation. The products from this reaction were unequivocally identified by X-ray crystallography. Through the use of DFT computational studies, the net 1,3-carboboration was shown to proceed via initial adduct formation followed by an aryl migration coupled with elimination of aryl iodide and finally, cyclisation. The generation of these aryl-substituted products in a metal-free fashion provides a facile route to boron containing heterocycles.
We are grateful to the EPSRC (T. A. G. P/L016443/1) and (R. L. M. EP/R026912/1) for funding and the awarding of an EPSRC Early Career Fellowship (R. L. M.). We also thank the Leverhulme Trust (D. W. and R. L. M. RPG-2016-020) and NSERC (J. M. R. DG 2015-05523) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. C. Brown, Compr. Organomet. Chem., 1982, 111–142 Search PubMed.
- M. Corpet and C. Gosmini, Synthesis, 2014, 2258–2271 CAS.
- J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 CAS.
- (a) T. Hackel and N. A. McGrath, Molecules, 2019, 24, 432–462 CrossRef PubMed; (b) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- D. W. Stephan, Science, 2016, 354, aaf7229-1–aaf7229-8 CrossRef PubMed.
- P. Laszlo and M. Teston, J. Am. Chem. Soc., 1990, 112, 8750–8754 CrossRef CAS.
- J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627–8643 CrossRef CAS PubMed.
- (a) B. Wrackmeyer, Heteroat. Chem., 2006, 17, 188–208 CrossRef CAS; (b) B. Wrackmeyer, Coord. Chem. Rev., 1995, 145, 125–156 CAS.
- G. Kehr and G. Erker, Chem. Sci., 2016, 7, 56–65 RSC.
- K. Bläsing, J. Bresien, R. Labbow, D. Michalik, A. Schulz, M. Thomas and A. Villinger, Angew. Chem., Int. Ed., 2019, 58, 6540–6541 CrossRef PubMed.
- (a) I. A. Cade and M. J. Ingleson, Chem. – Eur. J., 2014, 20, 12874–12880 CrossRef CAS PubMed; (b) M. Devilard, R. Brousses, K. Miqueu, G. Bouhadir and D. A. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5722–5726 CrossRef PubMed.
- R. L. Melen, L. C. Wilkins, B. M. Kariuki, H. Wadepohl, L. H. Gade, A. S. K. Hashmi, D. W. Stephan and M. M. Hansmann, Organometallics, 2015, 34, 4127–4137 CrossRef CAS.
- M. Mehta and J. M. Goicoechea, Chem. Commun., 2019, 55, 6918–6921 RSC.
- P. Hewavitharanage, E. O. Danilov and D. C. Neckers, J. Org. Chem., 2005, 70, 10653–10659 CrossRef CAS PubMed.
- (a) R. C. Neu, C. Jiang and D. W. Stephan, Dalton Trans., 2013, 42, 726–736 RSC; (b) R. C. Neu and D. W. Stephan, Organometallics, 2012, 31, 46–49 CrossRef CAS.
- M. Santi, D. M. C. Ould, J. Wenz, Y. Soltani, R. L. Melen and T. Wirth, Angew. Chem., Int. Ed., 2019, 58, 7861–7865 CrossRef CAS PubMed.
- M. S. Yusubov, A. Yoshimura and V. V. Zhdankin, Rev. Acc., 2016, 1, 342–374 Search PubMed.
- F. H. Allen, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, Int. Tables Crystallogr., 2006, 9.5(C), 790–811 Search PubMed.
- A. Nagai, K. Kokado, Y. Nagata, M. Arita and Y. Chujo, J. Org. Chem., 2008, 73, 8605–8607 CrossRef CAS PubMed.
- Q. Yin, Y. Soltani, R. L. Melen and M. Oestreich, Organometallics, 2017, 36, 2381–2384 CrossRef CAS.
- A. V. Manaev, T. A. Chibisova, K. A. Lyssenko, M. Yu. Antipin and V. F. Traven, Russ. Chem. Bull., 2006, 55, 2091–2094 CrossRef CAS.
- N. W. Alcock, A. P. Bozopoulos, E. Hatzigrigoriou and A. Varvoglis, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 1300–1303 CrossRef.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS.
Footnotes |
| † Information about the data that underpins the results presented in this article, including how to access them, can be found in the Cardiff University data catalogue at http://doi.org/10.17035/d.2019.0090828346. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra and details of the X-ray analyses of compounds. CCDC 1935886, 1935888, 1935890, 1936166 and 1963748. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc08749d |
| This journal is © The Royal Society of Chemistry 2020 |