
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fine-tuning thio-pyridazinediones as SMDC scaffolds (with intracellular thiol release via a novel self-immolative linker)†
Marcos
Fernández
 ,
André
Shamsabadi
and
Vijay
Chudasama
*
,
André
Shamsabadi
and
Vijay
Chudasama
*
Department of Chemistry, University College London, London, UK. E-mail: v.chudasama@ucl.ac.uk
First published on 18th December 2019
Abstract
Herein we report the synthesis of a library of thioalkyl- and thioaryl-pyridazinediones for thiol-based self-immolative release of cargo. A bisthioaryl-pyridazinedione is shown to be stable to serum protein albumin but unstable in intracellular conditions. A derivatised analogue underwent self-immolative degradation in cellular thiol conditions as evidenced by LC-MS/release of a turn-on fluorescence fluorophore; versatility of the thiol-pyridazinedione is demonstrated through synthesis of SMDC precursors that contain three different functional groups on the same central molecule.
Traditional chemotherapy relies on the premise that administration of highly potent cytotoxic drugs will preferentially neutralise rapidly dividing cells over normal ones.1 However, this form of therapy is invariably associated with a lack of specificity for tumours, often resulting in undesirable side effects. Furthermore, traditional low molecular weight cytotoxic agents utilised in such treatments display non-preferential or minimal accumulation at the tumour site in vivo,2–5 which has been explained by factors such as a tumour's irregular vasculature,6,7 the high interstitial pressure at the site,8 and the tumour's overexpression of multidrug-resistant proteins.9 In an effort to overcome the unfavourable characteristics associated with using conventional chemotherapy, important advances have been made in the past decades on tumour-targeted drug delivery systems (TTDDSs),1,2,10 which are designed to selectively deliver toxic cargoes to the tumour, thus reducing damage to healthy tissue and minimising dose-limiting side effects.
Whilst there has been significant focus on developing antibody–drug conjugates (ADCs) as TTDDSs, an alternative field that does not rely on protein-based targeting has emerged.11–13 In this approach, small organic ligands – such as binders of prostate-specific membrane antigen (PSMA), carbonic anhydrase IX, biotin and folates14–17 – act as the “vehicles” to selectively deliver a cytotoxic agent to tumours; this class of compounds are commonly termed small molecule–drug conjugates (SMDCs).2,5 The latter's advantages over, or complements to, protein-based approaches are manifold: their smaller size allows for faster and deeper tumour penetration, they have lower potential for immunogenic responses, are more easily synthesised, have lower manufacturing costs, and shorter distribution and elimination half-lives.2–4,14,18 Our lab has conducted research efforts into targeted therapy through the site-selective modification of antibodies,19 vNAR proteins20 and antibody fragments21 with bromopyridazinediones (BrPDs) that bear functional modalities. Site-selective attachment of these small molecules to protein cysteine(s) yields bioconjugates that are stable to hydrolysis and in serum-mimicking conditions, do not affect protein binding significantly and can be adapted for attachment to nanoparticles.22–24 As such, thus far, BrPDs have been extensively exploited for the site-selective modification of proteins in various contexts. In this work, we developed the pyridazinedione platform for application in an SMDC setting (Fig. 1), including the creation of a novel thiol-induced self-immolative linker that could be used for the attachment and subsequent controlled release of cargo.
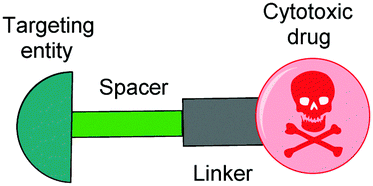 | ||
| Fig. 1 Schematic representation of an SMDC, typically consisting of a targeting ligand, a linker and/or spacer, and a cytotoxic drug payload. | ||
Two key considerations for an SMDC are that it is stable to blood thiols (primarily serum albumin) and is capable of releasing cargo in intracellular conditions through the action of a cleavable linker. From the outset, we wanted to exploit the fact that thio-substituted PDs can undergo substitution (via addition–elimination) in the presence of glutathione (GSH) 1; as GSH exists in high concentration intracellularly but in a low concentration extracellularly, it is often used as the trigger in the design of a cleavable linker. Thus, if the thiol groups on a thio-substituted PD could be manipulated into being self-immolative linkers, there exists an opportunity for an intracellular release mechanism of cargo if the thio-substituted PD is stable to reaction with serum albumin; there is also literature precedent on thiolate released self-immolative linkers.25–28
In view of the above, the balance between stability of the thio-substituted PDs to blood thiols whilst maintaining reactivity to intracellular GSH conditions was of great importance (Scheme 1). As such, our study began with the synthesis of mono-BrPD and di-BrPD (see ESI,† see Fig. S2 and S3), as conduits to generate and appraise a variety of aryl, alkyl and mixed aryl-alkyl thio-substituted PDs (Fig. 2). This selection was made in order to gauge which groups were more stable in serum and whether the mono- or di-substitution would significantly affect reactivity. The five thio-substituted PDs (2–6) were synthesised from the BrPDs in moderate to excellent yields without any optimisation (see ESI,† Fig. S4–S9).
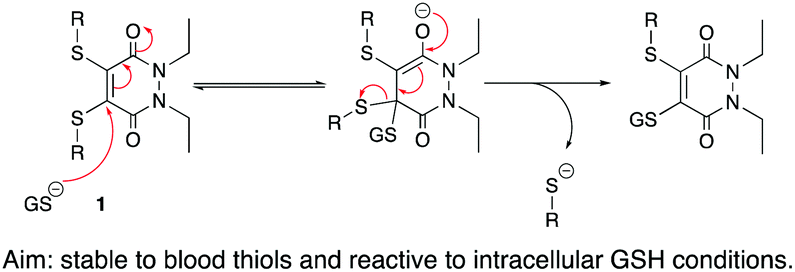 | ||
| Scheme 1 Addition–elimination mechanism under intracellular GSH conditions of thio-substituted PDs. Also applicable to mono-substituted PDs. | ||
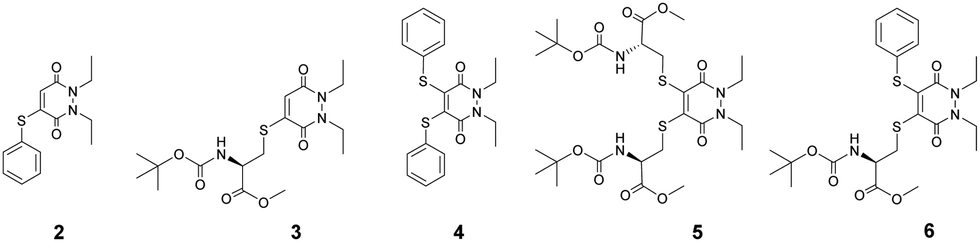 | ||
| Fig. 2 Structures of thio-substituted PDs 2–6. | ||
After the formation of PDs 2–6 (Fig. 2), we appraised their stability in serum-mimicking conditions. All five synthesised PDs 2–6 were incubated with human serum albumin (HSA), the blood's most abundant thiol, at pH 7.4 for 4 h, to determine if there was any reactivity with the protein's accessible cysteine residue. Gratifyingly, no reaction was observed between HSA and all of the trialled PDs, corroborating previously reported results that alkyl thio-PDs are stable in blood-mimicking conditions29 and unveiling aryl thio-PDs to also be stable in the time-frame of the experiment. As a positive control, maleimide was incubated with HSA alongside PDs 2–6 under identical conditions. Maleimides are substantially more reactive towards thiols than PDs and are known to react with HSA,30 therefore conjugation of HSA by maleimide was expected, and indeed observed. As some SMDCs, such as those based on folic acid for instance, have a blood circulation half-life of only 26 min,25 the substantially longer incubation time of 4 h demonstrates excellent stability of all the PDs in human blood serum-like conditions in this context.
We next explored the stability of PDs 2–6 in the presence of GSH 1, due to it being predominant in the intracellular milieu31,32 and its ability to cleave SMDCs to liberate free drug in other systems.10,31,33 It was hoped that PDs 2–6 would undergo an addition–elimination mechanism with GSH 1 to expel the thioaryl and thioalkyl moieties. As the concentration of intracellular GSH 1 falls in the range of 1 mM to 10 mM,31,34–36 an intermediate intracellular GSH concentration of 5 mM was considered appropriate for the study. Moreover, since the HSA studies employed an excess of PDs (10 eq.) to demonstrate their stability in blood, their expected instability when exposed to intracellular GSH called for GSH 1 to be used in excess (10 eq.), to mimic the large excess of this thiol inside a cell relative to the intracellular drug concentration. Thus, it was chosen for PDs 2–6 to be at a concentration of 0.5 mM relative to GSH 1. As many SMDCs are internalised by cells via receptor-mediated endocytosis (RME), the drug conjugate must necessarily pass through the early endosome before being degraded in the late endosome/lysosome. The pH extremes of the early endosome and late endosome/lysosome are around 6.5 and 4.9–5.0 respectively37–40 and thus it was decided to incubate PDs 2–6 with GSH 1 at pHs 6.5 and 5.0 with LC-MS readings taken at time points up to 24 h.
The studies at pH 6.5 were conducted first (see ESI,† Fig. S32–S56), as it was reasoned that if substitution were to take place, as was desired, it would have a greater chance of occurring at the higher of the endosome pH extremes. Bis-thioaryl PD 4 proved to be the superior candidate of all the five tested (Scheme 2). Reaction of PD 4 could give rise to two possible PD substitution products: mono-GSH PD and bis-GSH PD 7. After 4 h, only bis-GSH PD 7 was observed in the LC-MS trace, suggesting that the reaction had reached completion.
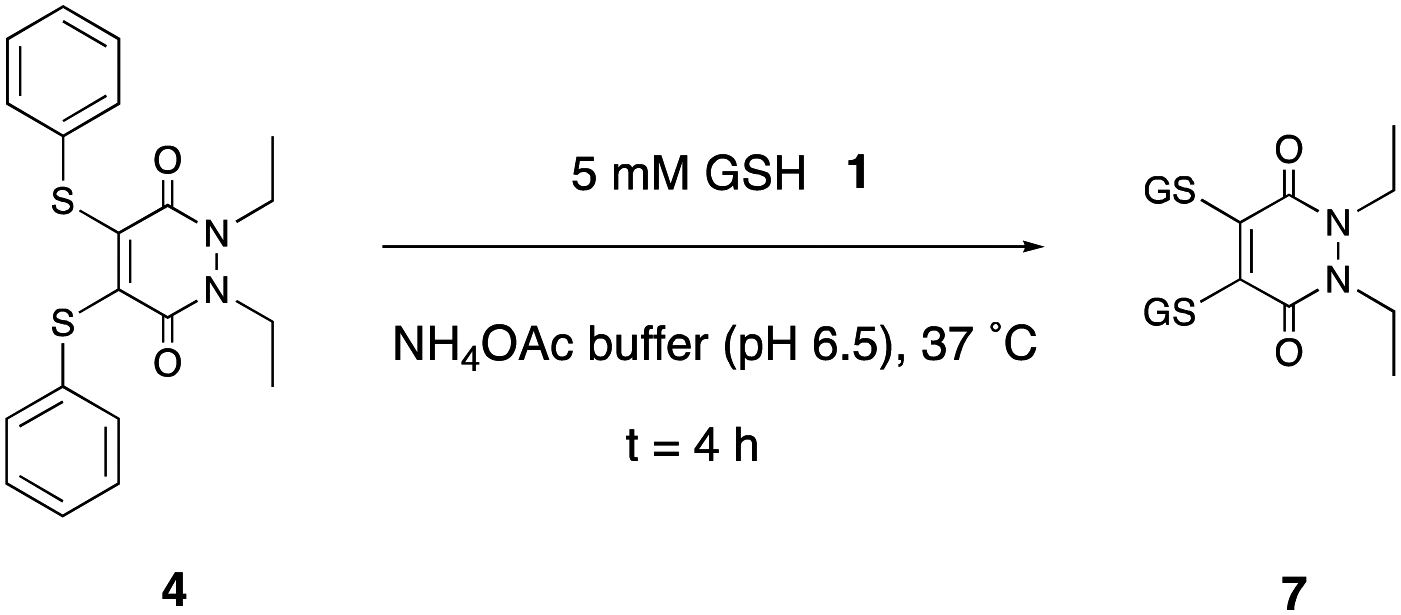 | ||
| Scheme 2 Incubation of PD 4 with GSH 1 to form PD 7 after 4 h. | ||
When all the studies were conducted at the lower pH extreme of 5.0, the consumption of 2, 3, 5 and 6 was negligible relative to that of PD 4 (see ESI,† Fig. S57–S81).
Both pH studies thus confirmed bis-thioaryl PD 4 as the best candidate of the five to be brought forward in derivatising the thioaryl moieties of its PD backbone for the purpose of creating a GSH-triggered cleavable, self-immolative linker. To do this, we considered the work of Van de Bittner et al., who developed a self-immolative phenolic-based construct that releases luciferin.41 Gois et al. modified this phenolic-based linker to release umbelliferone 12 upon self-immolative degradation.42 Taking inspiration from both of these systems, we decided to apply a related, but novel, strategy to the thiophenol system of PD 4, which required the synthesis of umbelliferone–PD 13. To this end, we synthesised PD 8, which could be brominated using N-bromosuccinimde (NBS) 9 and azobisisobutyronitrile (AIBN) 10 to make di-bromo intermediate 11. Displacement of the bromine groups with the phenolic functionality on umbelliferone 12, a commonly used “turn-on” fluorescent probe (not fluorescent when phenol is substituted with anything other than H), was then carried out to obtain umbelliferone–PD 13 (Scheme 3).
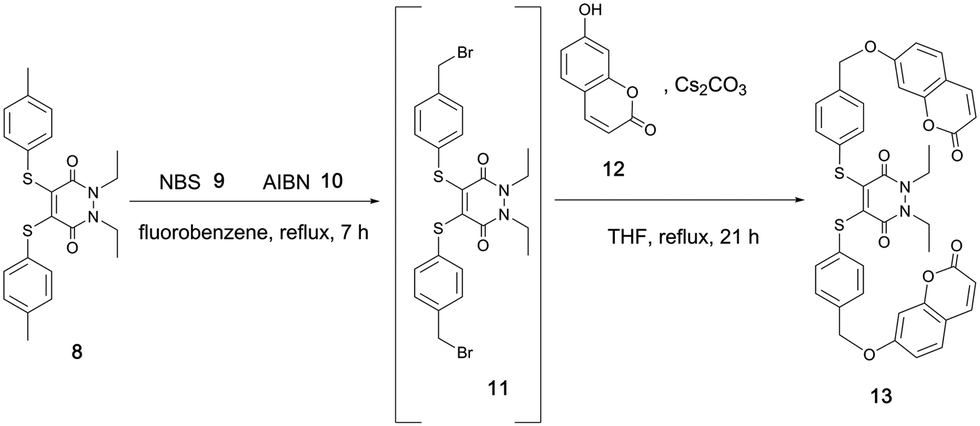 | ||
| Scheme 3 Synthesis of PD 13 which utilises two thioaryl-linked umbelliferone groups. | ||
As the GSH studies had confirmed that these intracellular thiol conditions did displace the aryl moieties of related PD 4, it was anticipated that subjecting umbelliferone–PD 13 to 5 mM GSH at pH 6.5 would displace its thioaryl groups in a similar manner. If so, thiophenol-based construct 14 should be expelled, which we hoped would undergo self-immolative cleavage to yield fluorescent umbelliferone 12 (Scheme 4). Gratifyingly, when umbelliferone–PD 13 was incubated with GSH 1 at pH 6.5 for 24 h, expulsion of umbelliferone 12 was observed by LC-MS, confirming the self-immolative mechanism based on thiophenol. It was also pleasing that construct 14 was not visible by LC-MS as this indicated that the self-immolative linker efficiently fragmented under the conditions.
 | ||
| Scheme 4 Reaction of umbelliferone–PD 13 with GSH 1 and proposed self-immolative breakdown of 14 to release fluorescent umbelliferone 12. | ||
Disappointingly however, a reaction suspension was observed upon incubation of PD 13 with GSH 1, suggesting that umbelliferone–PD 13 was not fully soluble in a THF/ammonium acetate buffer mixture; this may also have gone some way to explain the lack of full conversion of PD 13 after 24 h under the reaction conditions. As such, it was pertinent to tailor the structure of umbelliferone–PD 13 in an effort to make it more water-soluble.
It was envisaged that adding a carboxylic acid group to the 3-position of each umbelliferone moiety in umbelliferone–PD 13 would aid water solubility. To this end, 3-tert-butyl carboxylic acid umbelliferone was synthesised prior to base-promoted reaction with di-bromo species 11 to afford a tert-butyl protected umbelliferone–PD, which was then stirred in 25% TFA/CH2Cl2 at 21 °C for 4 h to yield the desired carboxylic acid–umbelliferone–PD 16 (see ESI,† Fig. S12–S14).
With carboxylic acid–umbelliferone–PD 16 in hand, it was then appropriate to repeat the incubation with GSH 1 at pH 6.5, to test whether the addition of the carboxylic acid groups to the structure aided water solubility. Gratifyingly, nearly full consumption of PD 16 had occurred after 4 h, with no solubility issues observed. The analogue of intermediate 14 was observed at t = 0.05 h in low levels, but was consumed by the 4 h time point. Moreover, the main peaks observed by LC-MS after the 4 h time point were bis-GSH PD 7 and the released fluorophore 17 (see ESI,† Fig. S84–S86). This provided confirmation that PD 16 has a similar reactivity profile to model PD 4, and that greater solubility was key to this correlation. As well as the aforementioned structures being observed by LC-MS, the fluorescence emission spectrum of the reaction was also monitored, with the fluorescence increasing sharply between the t = 0.05 h and 4 h time points, with a less significant increase between the t = 4 h and 24 h time points (Fig. 3). Taken together, the LC-MS and fluorescence emission data confirm the turn-on fluorescence nature of this reaction, which could have important applications in cell imaging.
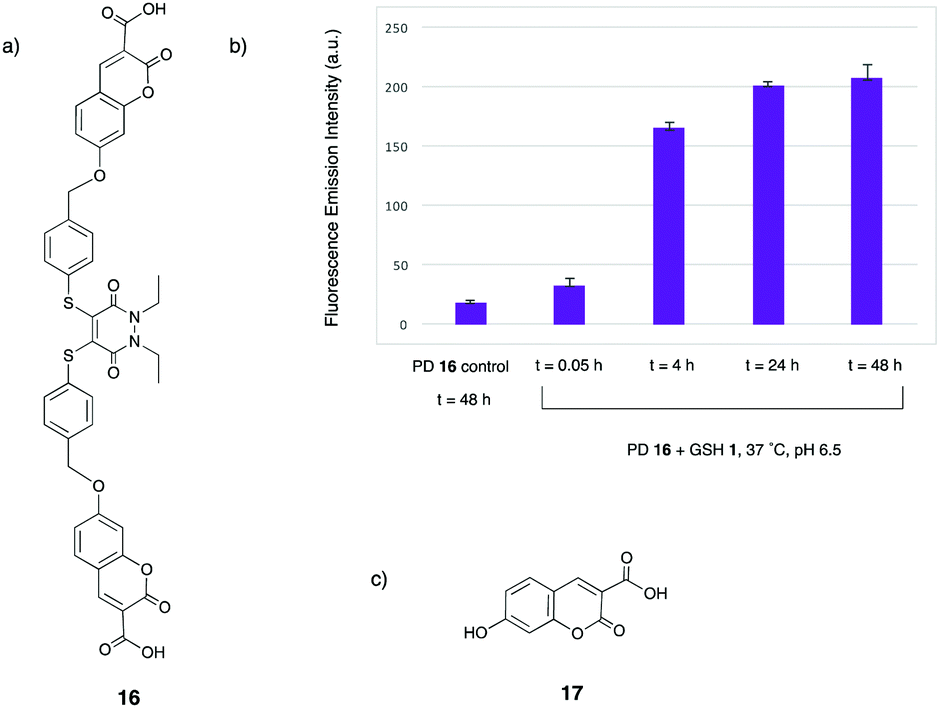 | ||
| Fig. 3 (a) Structure of carboxylic acid–umbelliferone–PD 16, (b) the turn-on fluorescence emission data for the incubation of PD 16 + GSH 1 at 37 °C at pH 6.5 and (c) structure of released fluorescent carboxylic acid–umbelliferone 17. | ||
With the self-immolative breakdown of a PD's thiolate moieties demonstrated, attention turned to substituting the N-alkyl portions of a BrPD into useful functional moieties. To this end an acid group and a simple PEG were chosen (Scheme 5).
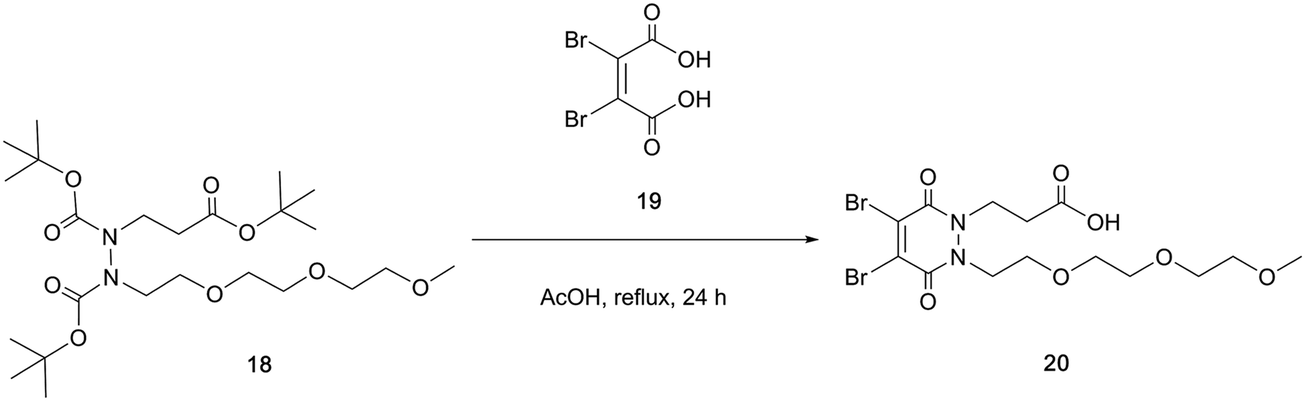 | ||
| Scheme 5 Synthesis of di-bromo PEG acid PD 20 from hydrazine 18 and dibromomaleic acid 19. | ||
To synthesise di-bromo PEG acid PD 20, PEG ester hydrazine 18 was synthesised using sequential alkylations of hydrazines followed by refluxing with dibromomaleic acid 19 to yield PD 20 in 74% yield. The reason for adding the PEG functionality was to aid solubility in order to counteract the attachment of any hydrophobic group that may be used in an SMDC (e.g. drug, fluorophore, porphyrin, etc.). The acid group can be readily functionalised with a plethora of commercially or readily available amine-bearing cargos that are relevant to SMDCs. This functionalisation was demonstrated by synthesising di-bromo PEG azide PD 21 and di-bromo PEG alkyne PD 22, as azide and alkyne groups are very useful for “click” modification. With these amine-functionalised PDs in hand, the final step in synthesising SMDC precursors involved substituting the bromine atoms for a derivatisable thiol, namely 4-methylbenzenethiol 23 (Scheme 6).
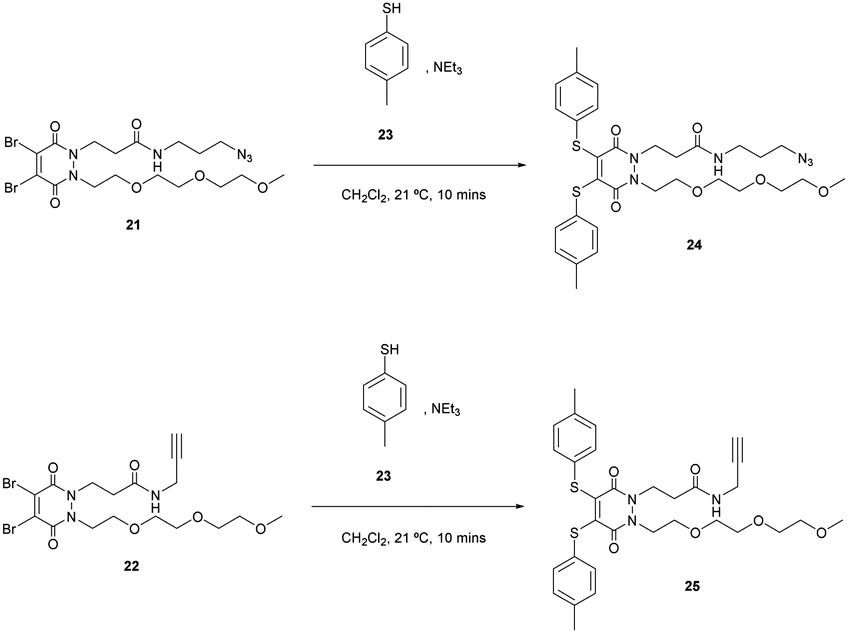 | ||
| Scheme 6 Syntheses of bis-thiol PEG azide and alkyne PDs 24 and 25. | ||
PDs 24 and 25 were obtained in 82% and 85% yield respectively. They constitute SMDC precursors where bromination of the methyl thiol (possibly after “click” modification) could be used for fluorophore/drug installation, and where the azide/alkyne groups serve as “click” handles for incorporating tumour-targeting ligands.
In conclusion, we have demonstrated the development of a novel, sulfur-based self-immolative linker that was utilised to incorporate a turn-on fluorescence fluorophore that is only released from the PD construct by GSH under conditions mimicking intracellular conditions, which in some cases is similar to certain tumour microenvironments. The first PD–fluorophore construct (13) suffered from suboptimal buffer solubility, and this issue was solved by installing two carboxylic groups onto the 3-position of the umbelliferone moieties to form PD–fluorophore 16. This construct displayed full solubility in the organic solvent/buffer mixture used, and the release of the fluorophore was measured in terms of mass by LC-MS, and the turn-on fluorescence nature of the reaction was monitored by fluorescence emission spectroscopy. Moreover, we developed azide- and alkyne-based SMDC precursors (24 and 25 respectively), with the PD scaffold's modularity and versatility shown by the installation of three different groups relevant to SMDCs on the same central molecule. The methyl portions of 24 and 25 are available for derivatisation in order to install fluorophores/drugs, whereas the azide and alkyne functionalities can be employed for the attachment of tumour-targeting ligands via “click” reactions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. D. Seitz, J. G. Vineberg, L. Wei, J. F. Khan, B. Lichtenthal, C. F. Lin and I. Ojima, J. Fluorine Chem., 2015, 171, 148–161 CrossRef CAS PubMed.
- N. Krall, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2013, 52, 1384–1402 CrossRef CAS PubMed.
- N. Krall, F. Pretto and D. Neri, Chem. Sci., 2014, 5, 3640–3644 RSC.
- A. Dal Corso, D. Neri and S. Cazzamalli, J. Controlled Release, 2017, 246, 39–45 CrossRef PubMed.
- G. Casi and D. Neri, Mol. Pharmaceutics, 2015, 12, 1880–1884 CrossRef CAS PubMed.
- P. Baluk, S. Morikawa, A. Haskell, M. Mancuso and D. M. McDonald, Am. J. Pathol., 2003, 163, 1801–1815 CrossRef PubMed.
- E. di Tomaso, D. Capen, A. Haskell, J. Hart, J. J. Logie, R. K. Jain, D. M. McDonald, R. Jones and L. L. Munn, Cancer Res., 2005, 65, 5740–5749 CrossRef CAS PubMed.
- V. P. Chauhan, T. Stylianopoulos, Y. Boucher and R. K. Jain, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 281–298 CrossRef CAS PubMed.
- R. O’Connor, Anticancer Res., 2007, 27, 1267–1272 Search PubMed.
- J. G. Vineberg, E. S. Zuniga, A. Kamath, Y. J. Chen, J. D. Seitz and I. Ojima, J. Med. Chem., 2014, 57, 5777–5791 CrossRef CAS PubMed.
- C. Zhuang, X. Guan, H. Ma, H. Cong, W. Zhang and Z. Miao, Eur. J. Med. Chem., 2019, 163, 883–895 CrossRef CAS PubMed.
- G. Casi and D. Neri, J. Med. Chem., 2015, 58, 8751–8761 CrossRef CAS PubMed.
- M. Srinivasarao, C. V. Galliford and P. S. Low, Nat. Rev. Drug Discovery, 2015, 14, 203–219 CrossRef CAS PubMed.
- N. Krall, F. Pretto, W. Decurtins, G. J. L. Bernardes, C. T. Supuran and D. Neri, Angew. Chem., Int. Ed., 2014, 53, 4231–4235 CrossRef CAS PubMed.
- S. A. Kularatne, K. Wang, H. K. Santhapuram and P. S. Low, Mol. Pharmaceutics, 2009, 6, 780–789 CrossRef CAS PubMed.
- S. Bhuniya, S. Maiti, E. J. Kim, H. Lee, J. L. Sessler, K. S. Hong and J. S. Kim, Angew. Chem., Int. Ed., 2014, 53, 4469–4474 CrossRef CAS PubMed.
- C. Chen, J. Ke, X. Edward Zhou, W. Yi, J. S. Brunzelle, J. Li, E. L. Yong, H. E. Xu and K. Melcher, Nature, 2013, 500, 486–489 CrossRef CAS PubMed.
- A. Kumar, T. Mastren, B. Wang, J. T. Hsieh, G. Hao and X. Sun, Bioconjugate Chem., 2016, 27, 1681–1689 CrossRef CAS PubMed.
- E. Robinson, J. P. M. Nunes, V. Vassileva, A. Maruania, J. C. F. Nogueira, M. E. B. Smith, R. B. Pedley and S. Caddick, RSC Adv., 2017, 7, 9073–9077 RSC.
- J. C. F. Nogueira, M. K. Greene, D. A. Richards, A. O. Furby, J. Steven, A. Porter, C. Barelle, C. J. Scott and V. Chudasama, Chem. Commun., 2019, 55, 7671–7674 RSC.
- M. K. Greene, D. A. Richards, J. C. F. Nogueira, K. Campbell, P. Smyth, M. Fernandez, C. J. Scott and V. Chudasama, Chem. Sci., 2018, 9, 79–87 RSC.
- A. Maruani, H. Savoie, F. Bryden, S. Caddick, R. Boyle and V. Chudasama, Chem. Commun., 2015, 51, 15304–15307 RSC.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645–6654 CrossRef CAS PubMed.
- A. Maruani, S. Alom, P. Canavelli, M. T. W. Lee, R. E. Morgan, V. Chudasama and S. Caddick, Chem. Commun., 2015, 51, 5279–5282 RSC.
- I. R. Vlahov and C. P. Leamon, Bioconjugate Chem., 2012, 23, 1357–1369 CrossRef CAS PubMed.
- C. F. Riber, A. A. A. Smith and A. N. Zelikin, Adv. Healthcare Mater., 2015, 4, 1887–1890 CrossRef CAS PubMed.
- M. Sengee, J. J. Eksteen, S. L. Nergard, T. Vasskog and L. K. Sydnes, Bioconjugate Chem., 2019, 30, 1489–1499 CrossRef CAS PubMed.
- M. Gund, A. Khanna, N. Dubash, A. Damre, K. S. Singh and A. Satyam, Bioorg. Med. Chem. Lett., 2015, 25, 122–127 CrossRef CAS PubMed.
- E. Robinson, J. P. M. Nunes, V. Vassileva, A. Maruani, J. C. F. Nogueira, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 9073–9077 RSC.
- M. E. B. Smith, M. B. Caspersen, E. Robinson, M. Morais, A. Maruani, J. P. M. Nunes, K. Nicholls, M. J. Saxton, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2015, 13, 7946–7949 RSC.
- D. Feng, Y. Song, W. Shi, X. Li and H. Ma, Anal. Chem., 2013, 85, 6530–6535 CrossRef CAS PubMed.
- M. H. Lee, J. Y. Kim, J. H. Han, S. Bhuniya, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 12668–12674 CrossRef CAS PubMed.
- J. D. Seitz, J. G. Vineberg, E. Herlihy, B. Park, E. Melief and I. Ojima, Bioorg. Med. Chem., 2015, 23, 2187–2194 CrossRef CAS PubMed.
- J. X. Lu, Y. C. Song, W. Shi, X. H. Li and A. H. Maurer, Sens. Actuators, B, 2012, 161, 615–620 CrossRef CAS.
- S. J. Wang, H. M. Ma, J. Li, X. Q. Chen, Z. J. Bao and S. N. Sun, Talanta, 2006, 70, 518–521 CrossRef CAS PubMed.
- S. Santra, C. Kaittanis, O. J. Santiesteban and J. M. Perez, J. Am. Chem. Soc., 2011, 133, 16680–16688 CrossRef CAS PubMed.
- Y.-B. Hu, E. B. Dammer, R.-J. Ren and G. Wang, Transl. Neurodegener., 2015, 4, 1–10 CrossRef PubMed.
- G. H. Diering and M. Numata, Front. Physiol., 2014, 4, 1–7 Search PubMed.
- J. Huotari and A. Helenius, EMBO J., 2011, 30, 3481–3500 CrossRef CAS PubMed.
- M. J. Geisow and W. H. Evans, Exp. Cell Res., 1984, 150, 36–46 CrossRef CAS PubMed.
- G. C. Van de Bittner, E. A. Dubikovskaya, C. R. Bertozzi and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21316–21321 CrossRef CAS PubMed.
- F. M. F. Santos, A. I. Matos, A. E. Ventura, J. Gonçalves, L. F. Veiros, H. F. Florindo and P. M. P. Gois, Angew. Chem., Int. Ed., 2017, 56, 9346–9350 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc08744c |
| This journal is © The Royal Society of Chemistry 2020 |