
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHomocoupling of CO and isocyanide mediated by a C,C′-bis(silylenyl)-substituted ortho-carborane†
Yun
Xiong
 a,
Shenglai
Yao
a,
Tibor
Szilvási
b,
Ales
Ruzicka
c and
Matthias
Driess
*a
a,
Shenglai
Yao
a,
Tibor
Szilvási
b,
Ales
Ruzicka
c and
Matthias
Driess
*a
aTechnische Universität Berlin, Institute of Chemistry: Metalorganic and Inorganic Materials, Sekr. C2, Strasse des 17. Juni 135, 10623 Berlin, Germany. E-mail: matthias.driess@tu-berlin.de; Fax: +49-30-314-29732
bDepartment of Chemical & Biological Engineering University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA
cDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentska 573, 532 10 Pardubice, Czech Republic
First published on 10th December 2019
Abstract
The unexpected reactivity of the o-carborane supported bis-silylene [(LSi:)C]2B10H101 {L= PhC(tBuN)2} towards carbon monoxide and 2,6-dimethylphenyl isocyanide is reported. While the reaction of 1 with CO leads selectively to the novel head-to-head coupling and C–O cleavage product 2 from two molecules 1 and four molecules CO, the reaction of 1 with 2,6-dimethylphenyl isocyanide affords solely the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar head-to-tail coupling product 3 with a Si
:2 molar head-to-tail coupling product 3 with a Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.
C bond.
The activation of important small molecules such as CO, H2 and N2 was earlier considered to be a domain of transition metals. However, the last decade witnessed increasing achievements in the chemistry of main-group element (MG) compounds that mediate the cleavage of strong bonds in small molecules.1–7 Featuring a lone pair of electrons and a vacant p-orbital at the silicon center, silylenes are particularly reactive species which belong to the rare examples of low-valent MG compounds being suitable for the activation of unreactive bonds and thus represent versatile building blocks for the synthesis of value-added organosilanes. Since the isolation of the first isolable N-heterocyclic silylene (NHSi) by West et al. in 1994,8 the chemistry of stable silylenes has flourished substantially. Several cyclic9–13 and acyclic14–18 silylenes have been successfully synthesised and their reactivity towards numerous small molecules such as O2, N2O, S8, P4, NH3, CO2, and even H2 has been developed.5,15–17,19 Nevertheless, the very strong triple bonds of CO cannot be activated easily; one very recent example represents the homocoupling of CO molecules to OCCO moieties mediated by an acyclic monosilylene reported by Aldridge and co-workers.20
In order to learn whether two silylene (NHSi) moieties could be cooperative in cleaving unreactive bonds, we synthesised several chelating bis-NHSis.21–24 To our delight, preliminary investigations revealed that the xanthene and ferrocene supported bis-NHSi A23a and B21b can even cleave the strong C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O bond (1072 kJ mol−1) and mediates the deoxygenative coupling of two CO molecules under mild reaction conditions (room temperature, 1 atm) to yield the ketene species C and D, respectively (Scheme 1).23b,c Moreover, the reaction of bis-silylene A with an equimolar amount of the isoelectronic 2,6-dimethylphenyl isocyanide resulted in the dearomatised silene derivative E.23b Moreover, the reaction of A with two molar equivalents of 2,6-dimethylphenyl isocyanide afforded F and G.
O bond (1072 kJ mol−1) and mediates the deoxygenative coupling of two CO molecules under mild reaction conditions (room temperature, 1 atm) to yield the ketene species C and D, respectively (Scheme 1).23b,c Moreover, the reaction of bis-silylene A with an equimolar amount of the isoelectronic 2,6-dimethylphenyl isocyanide resulted in the dearomatised silene derivative E.23b Moreover, the reaction of A with two molar equivalents of 2,6-dimethylphenyl isocyanide afforded F and G.
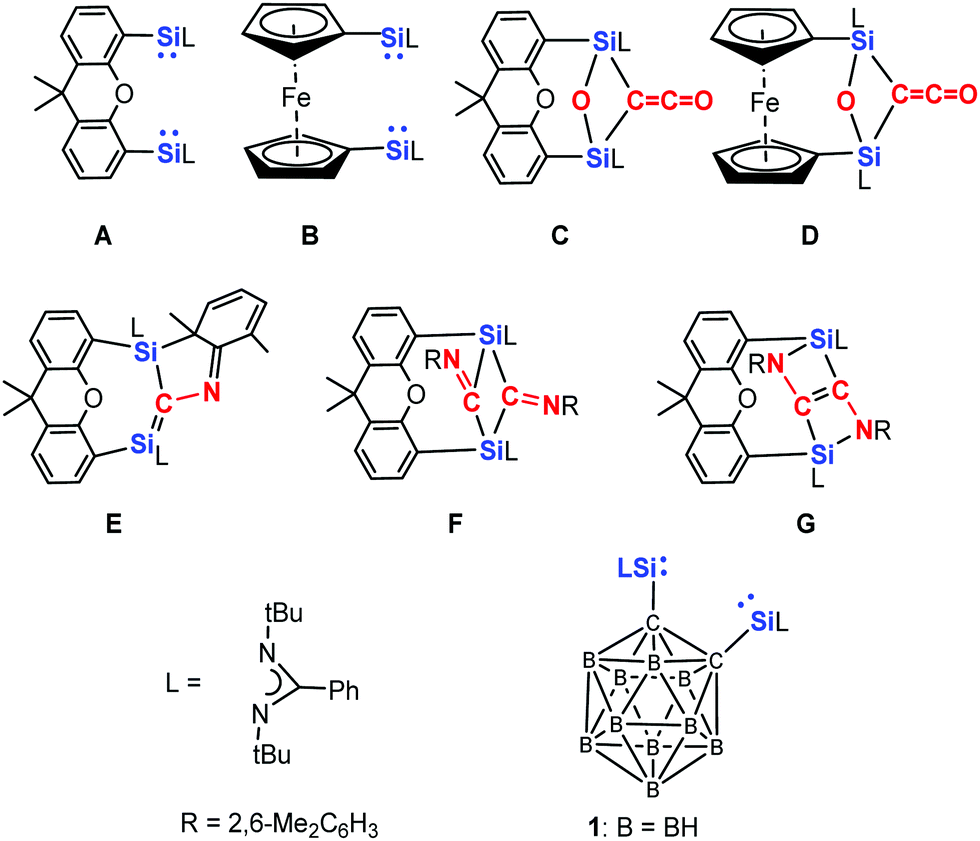 | ||
| Scheme 1 Bis-silylenes A, B and 1 as well as the products C–G resulting from the reaction of A and B with CO and 2,6-dimethylphenyl isocyanide, respectively. | ||
Compared to the bis-silylene A,23a the ortho-carborane supported bis-NHSi 124 is more rigid, and we wondered its reactivity towards CO and the isoelectronic 2,6-dimethylphenyl isocyanide furnishes similar or different coupling products. Herein, we report the unexpected novel coupling products from CO and 2,6-dimethylphenyl isocyanide mediated by the bis(NHSi) 1.
Exposure of the yellow solution of 1 in toluene to CO (1 atm) at −20 °C leads to a colour change from yellow to brown. The reaction mixture was allowed to warm to room temperature to give a brown solution, from which the unprecedented coupling compound 2 could be crystallised after one day at ambient temperature in the form of colourless crystals in 62% yields (Scheme 2). In contrast to the reaction of A and B with CO, the corresponding disilaketene could even not be observed as intermediates, presumably owing to the rigidity and steric hindrance of the o-carborane backbone.
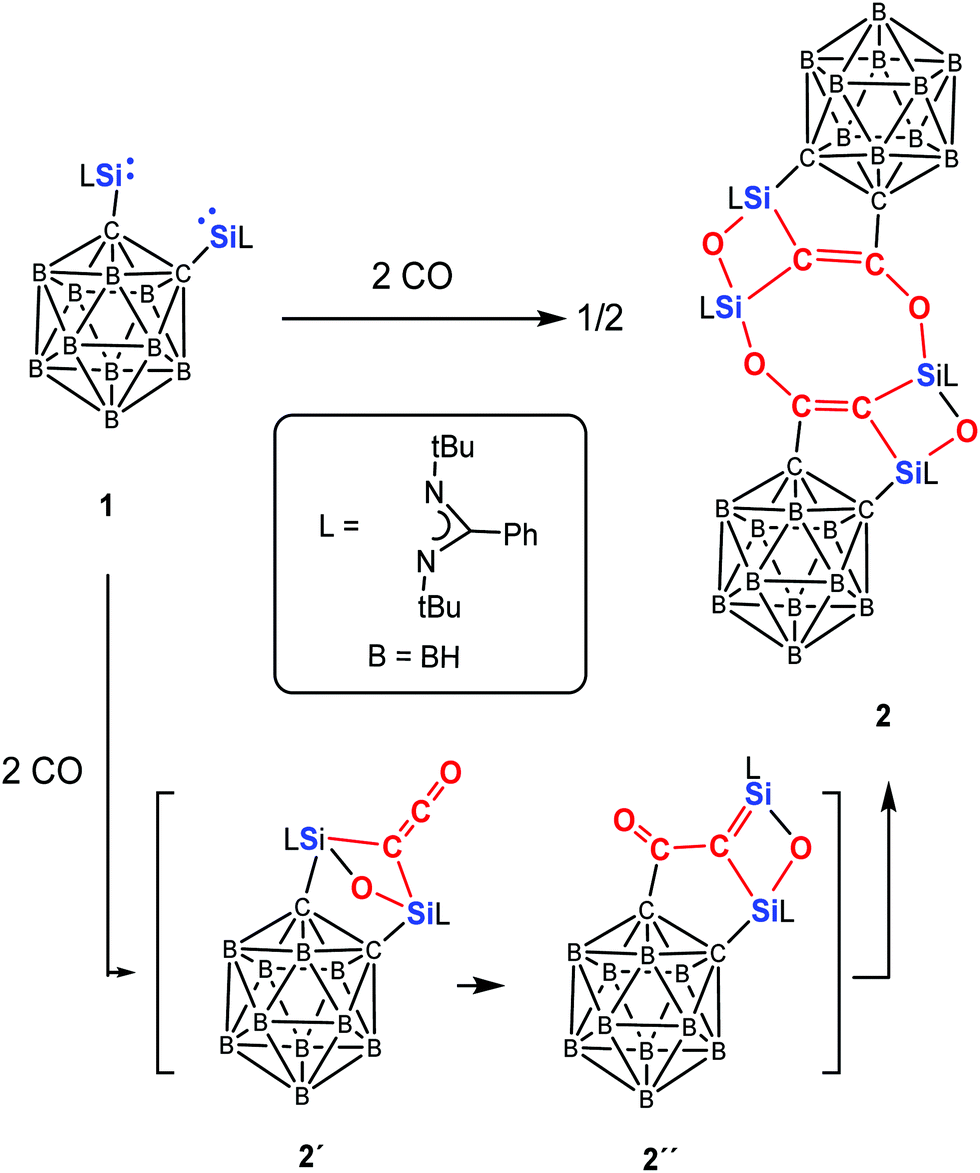 | ||
| Scheme 2 Reaction of bis-silylene 1 with CO to afford 2via2′ and 2′′. | ||
In compound 2, the two different sorts of 29Si nuclei give rise to different 29Si NMR resonances. The resonance at δ = −62.27 ppm can be assigned to the Si atoms of the CSiO2 moiety, whereas that at δ = −108.44 ppm corresponds to the C2SiO units; the latter value is close to the 29Si NMR resonance of C (δ = −91.4 ppm).23b Interestingly, the CC–O moieties in 2 resonate at 22.34 and 173.78 ppm in the 13C NMR spectrum, which are reminiscent of those observed for the ketene moiety in C (19.7 and 165.5 ppm).
A single crystal X-ray diffraction analysis revealed a polycyclic skeleton in 2 as depicted in Fig. 1. Compound 2 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Its molecular structure consists of an eight-membered Si2C4O2 ring and two four-membered SiOSiC rings. The penta-coordinate Si1 and Si3 atoms in the eight-membered ring feature a distorted trigonal bipyramidal coordination geometry with the N1–Si1–C3 angle of 172.2(1) and the N6–Si3–C6 angle of 170.6(1)° in the axial direction, respectively. In comparison, the Si2 and Si4 atoms are also penta-coordinated (ON2C2), but adopt a stronger distorted trigonal bipyramidal geometry with the C8–Si4–O3 angle of 162.1(1) and N3–Si2–C3 of 161.3(1)°, respectively. The Si–O distances, ranging from 1.683(3) to 1.728(2) Å, are in the common range of Si–O bonds. The Si–C distances with the carborane C atoms (Si2–C1 2.042(3) and Si4–C8 2.070(3) Å) are significantly longer than those of the C atoms in the eight-membered Si2C4O2 ring (1.876(3) to 1.905(3) Å). The C4–C2 (1.537(3) Å) and C5–C7 (1.536(4) Å) interactions, bridging the eight-membered ring and the two carborane clusters, represent C–C single bonds. However, the C–C distances within the eight-membered ring C3–C4 (1.324(4) Å) and C5–C6 (1.337(4) Å) are significantly shorter and indicative of CC bonds. It is worthy of note that the C4–O4 (1.361(3) Å) and C5–O2 bonds (1.346(4) Å) are much shorter than a C–O bond in ethers, implying considerable electron delocalisation in those C–C–O moieties.
. Its molecular structure consists of an eight-membered Si2C4O2 ring and two four-membered SiOSiC rings. The penta-coordinate Si1 and Si3 atoms in the eight-membered ring feature a distorted trigonal bipyramidal coordination geometry with the N1–Si1–C3 angle of 172.2(1) and the N6–Si3–C6 angle of 170.6(1)° in the axial direction, respectively. In comparison, the Si2 and Si4 atoms are also penta-coordinated (ON2C2), but adopt a stronger distorted trigonal bipyramidal geometry with the C8–Si4–O3 angle of 162.1(1) and N3–Si2–C3 of 161.3(1)°, respectively. The Si–O distances, ranging from 1.683(3) to 1.728(2) Å, are in the common range of Si–O bonds. The Si–C distances with the carborane C atoms (Si2–C1 2.042(3) and Si4–C8 2.070(3) Å) are significantly longer than those of the C atoms in the eight-membered Si2C4O2 ring (1.876(3) to 1.905(3) Å). The C4–C2 (1.537(3) Å) and C5–C7 (1.536(4) Å) interactions, bridging the eight-membered ring and the two carborane clusters, represent C–C single bonds. However, the C–C distances within the eight-membered ring C3–C4 (1.324(4) Å) and C5–C6 (1.337(4) Å) are significantly shorter and indicative of CC bonds. It is worthy of note that the C4–O4 (1.361(3) Å) and C5–O2 bonds (1.346(4) Å) are much shorter than a C–O bond in ethers, implying considerable electron delocalisation in those C–C–O moieties.
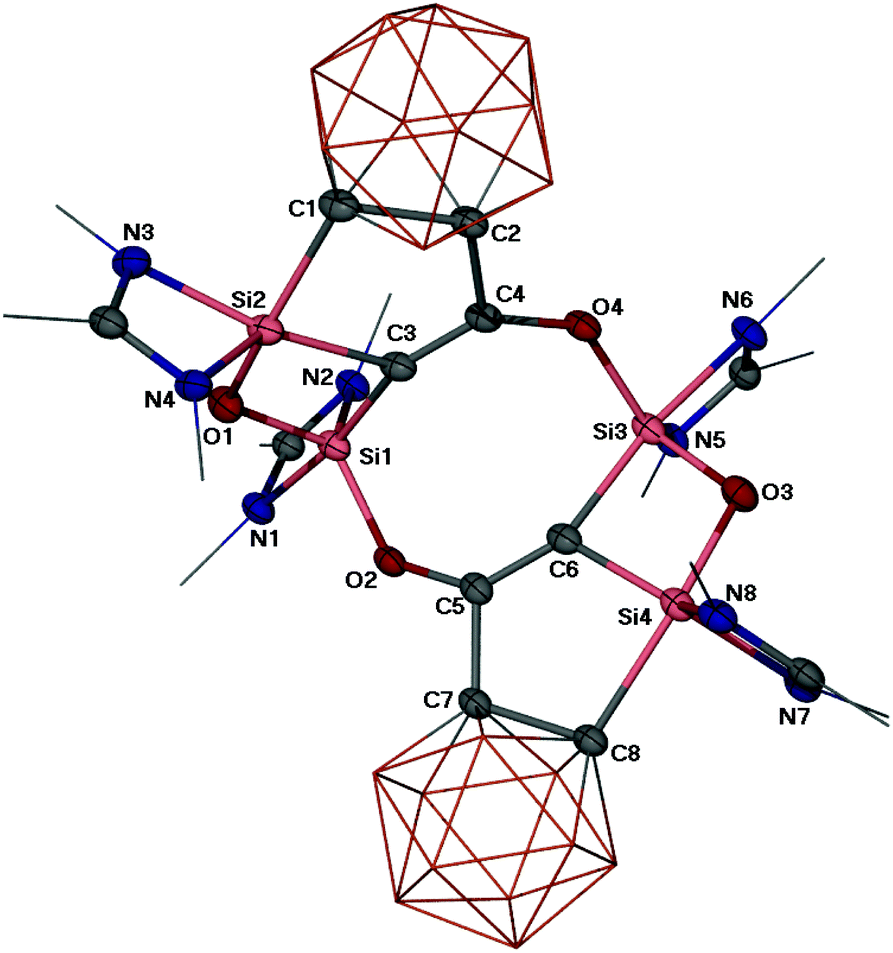 | ||
| Fig. 1 Molecular structure of 2. Thermal ellipsoids are drawn at 50% probability level. Hydrogen atoms and Ph as well as t-Bu groups are omitted for clarity. Selected bond distances (Å) and angles (°): Si1–O1 1.690(1), Si2–O1 1.698(2), Si1–O2 1.728(2), Si3–O3 1.683(3), Si4–O3 1.705(2), Si3–O4 1.724(2), Si1–C3 1.876(3), Si2–C3 1.905(3), C3–C4 1.324(4), C4–C2, 1.537(3), O4–C4 1.361(3), Si3–C6 1.878(3), Si4–C6 1.905(4), C5–C6 1.337(4), C5–O2 1.346(4), C5–C7 1.536(4), Si2–C1 2.042(3), Si4–C8 2.070(3),; Si1–O1–Si2 104.1(1), Si1–C3–Si2 89.9(1), Si3–O3–Si4 103.9(1), Si3–C6–Si4 89.7(1), C3–C4–O4 132.0(2), C6–C5–O2 132.2(3), C3–Si1–N1 172.2(1), C3–Si2–N3 161.3(1), N6–Si3–C6 170.6(1), C8–Si4–O3 162.1(1). | ||
The reactivity of 1 towards the isoelectronic 2,6-dimethylphenyl isocyanide has also been investigated (Scheme 3). 1H NMR measurements showed that compound 3 is formed as sole product independent of the molar ratio of the starting materials chosen. Thus reaction of 1 with two molar equivalents of isocyanide at −20 °C in toluene affords 3 in 78% isolated yields. This is in stark contrast to the reactions of bis-NHSi A with the same isocyanide,23b which led to E, F, and G (Scheme 1) as products depending on the molar ratio of the starting materials and the reaction conditions.
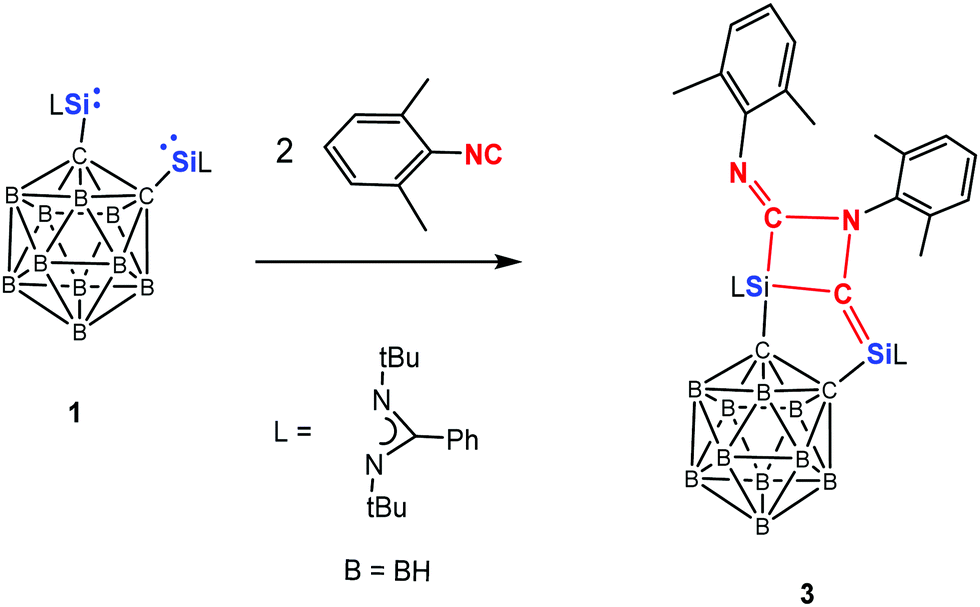 | ||
| Scheme 3 Reaction of 1 with 2,6-dimethylphenylisocyanide to form 3. | ||
The 29Si NMR spectrum of 3 shows two resonances at δ = −32.3 ppm (SiC) and −60.0 ppm (SiC3N2), assignable to the four- and five-coordinate Si atoms, respectively. The 13C{1H} NMR spectrum exhibits a signal at δ = 181.7 ppm for SiC–Si and 190.4 ppm for R–NC–Si (R = 2,6-dimethylphenyl), respectively. A single-crystal X-ray diffraction analysis revealed that 3 crystallises in the monoclinic space group P21/n. The molecular structure of 3 (Fig. 2) shows that the two molecules isocyanide underwent NC–N–C coupling and concomitantly Si–C bond formation. While the four-coordinate Si2 atom features a distorted tetrahedral coordination environment, the five-coordinate Si1 atom adopts a distorted trigonal bipyramidal coordination geometry with the axial C2–Si1–C5 angle of 165°. Both of the four-membered SiC2N and the five-membered Si2C3 rings are planar. Along the axial direction, the Si1–C2 (2.027(2) Å) and Si1–C5 (2.054(2) Å) distances are significantly longer than that of Si1–C3 (1.822(2) Å) in the equatorial direction. Moreover, the five-coordinate Si1 atom features also a longer Si1–C5 distance (2.054(2) Å) than that of the four-coordinate Si2 atom in the Si2–C6 bond (1.905(2) Å). The shortest Si2–C3 distance (1.724(2) Å) among all Si–C distances in 3 lies even in the range of SiC bond distances of silaethylene.25 Similarly, the relatively short C2–N6 (1.281(2) Å) distance compared with that of C3–N5 (1.474(2) Å) suggests CN bond character. It is noteworthy that compound 3 represents an unique example of head-to-tail coupling product of isocyanide compared to those usual head-to-head coupling products.26
 | ||
| Fig. 2 Molecular structure of 3. Thermal ellipsoids are drawn at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Si1–C3 1.822(2), Si1–C2 2.027(2), Si1–C5 2.054(2), Si2–C3 1.724(2), Si2–C6 1.905(2), C2–N6 1.281(2), C2–N5 1.413(2), C3–N5 1.474(2); C3–Si1–C2 70.7(1), C3–Si1–C5 94.3(1), C2–Si1–C5 165.0(1), C3–Si2–C6 101.6(1), N5–C3–Si2 139.9(1), N5–C3–Si1 97.3(1), Si2–C3–Si1 22.3(1). | ||
Because all attempts to detect an intermediate even at −78 °C by means of multi nuclear NMR spectroscopy were unsuccessful, Density Functional Theory (DFT) calculations were performed to gain insight into the mechanism of the formation of 2 and 3. The DFT-derived mechanism leading to 2 (Scheme 2 and Fig. S3 in ESI†) follows initially similar reaction steps that have been uncovered previously for the reaction of CO and bis-NHSi A to give C (Scheme 1).23b In contrast to the former case, however, the disilaketene analogue of C (compound 2′, Scheme 2) is a reactive intermediate which undergoes migration of one of the carbons of the carborane from Si to the central carbon of the ketene moiety to afford another intermediate (compound 2′′) with a SiC–CO moiety; head-to-tail dimerisation of the latter furnishes the final product 2. We think that the newly observed cleavage of the Ccarborane–Si bond is the consequence of the weaker and more polar nature of the C–Si bond as indicated by the lower Wiberg Bond Index (WBI, 0.53 in 1) compared to the WBI of 0.69 in both A and B. In addition, the proposed ketene intermediate 2′ is energetically 32.5 kcal mol−1 less stable than its dimer 2 in part owing to the steric congestion of 2′ caused by both bridged μ-CCO and μ-O. In case of the reaction of 1 with 2,6-dimethylphenyl isocyanide, the isocyanide moiety reacts rapidly with 1 to form 3′ which is attacked by an additional 2,6-dimethylphenyl isocyanide molecule to give 3 (Fig. 3). The latter reaction step shows a very small energy barrier of 1.9 kcal mol−1 that is consistent with the experimental fact that the conversion proceeds at −20 °C. NBO analysis (Tables S5–S7, ESI†) reveals the zwitterionic nature of 3′ (Fig. 3) that enables the reactivity of 3′ towards an additional isocyanide.
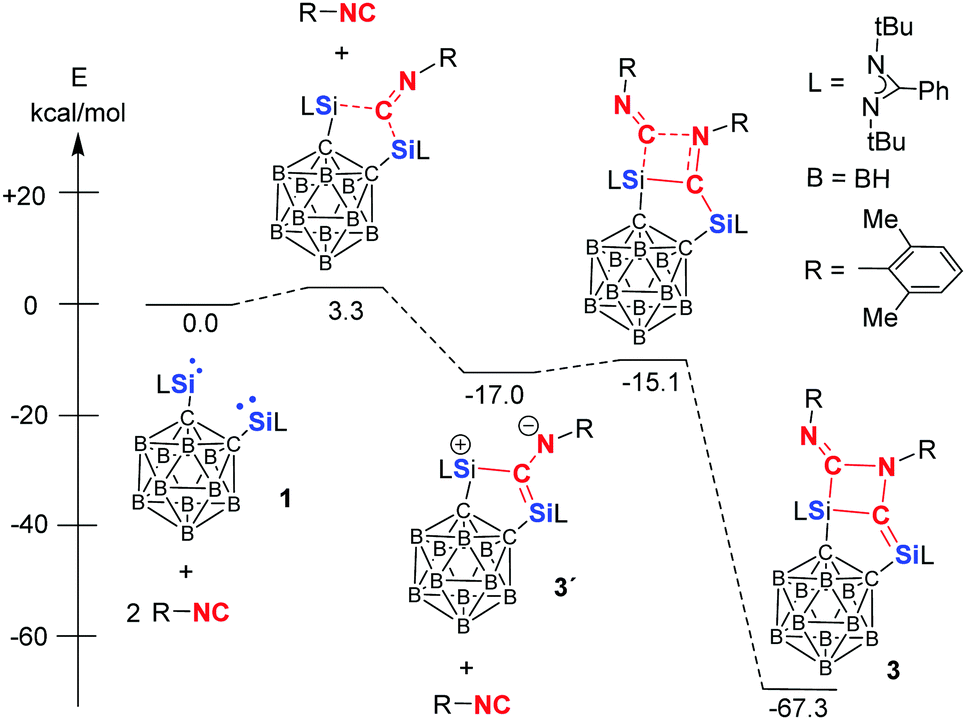 | ||
| Fig. 3 DFT-derived mechanism for the formation 3 from 1via3′. | ||
In conclusion, the o-carborane supported bis-NHSi 1 is more reactive towards CO and 2,6-dimethylphenyl isocyanide than the previously reported bis-NHSis A and B.23b Owing to the different steric and electronic effects of the o-carborane backbone, the outcome of the reaction of 1 with CO affords solely, via a cascade of cleavage and coupling reactions, the new bis-silylene-mediated polycyclic CO coupling product 2 as the final product. The reaction of 1 with 2,6-dimethylphenyl isocyanide furnishes the new SiC containing coupling product 3, irrespective of the chosen molar ratio of 1 to isocyanide. The unexpected head-to-head of CO and head-to-tail of isocyanide homocoupling products represent new reactivity features of a bis-NHSi towards small molecules. More investigations concerning the remarkable reactivity of bis-NHSi 1 to furnish new classes of organosilicon compounds are currently in progress.
We thank the Deutsche Forschungsgemeinschaft [DR 226/19-4] for financial support. Support of A. R. from the Czech Science Foundation (grant no. 17-10377S) is acknowledged.
Conflicts of interest
There are no conflicts to declare.References
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- S. Yadav, S. Saha and S. S. Sen, ChemCatChem, 2016, 8, 486–501 CrossRef CAS.
- D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC.
- P. P. Power, Chem. Rec., 2012, 12, 238–255 CrossRef CAS.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- (a) M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed; (b) M.-A. Légaré, M. Rang, G. Bélanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef PubMed.
- (a) X. Wang, Z. Zhu, Y. Peng, H. Lei, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2009, 131, 6912–6913 CrossRef CAS PubMed; (b) H. Braunschweig, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, J. O. C. Jimenez-Halla, T. Kramer, I. Krummenacher, J. Mies, A. K. Phukan and A. Vargas, Nat. Chem., 2013, 5, 1025–1028 CrossRef CAS PubMed.
- M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691–2692 CrossRef CAS.
- B. Gehrhus, M. F. Lappert, J. Heinicke, R. Boesec and D. Bläser, J. Chem. Soc., Chem. Commun., 1995, 1931–1932 RSC.
- M. Kira, S. Ishida, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 1999, 121, 9722–9723 CrossRef CAS.
- M. Driess, S. Yao, M. Brym, C. Van Wüllen and D. Lentz, J. Am. Chem. Soc., 2006, 128, 9628–9629 CrossRef CAS PubMed.
- C.-W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., Int. Ed., 2006, 45, 3948–3950 CrossRef CAS PubMed.
- M. Asay, S. Inoue and M. Driess, Angew. Chem., Int. Ed., 2011, 50, 9589–9592 CrossRef CAS PubMed.
- B. D. Rekken, T. M. Brown, J. C. Fettinger, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2012, 134, 6504–6507 CrossRef CAS PubMed.
- A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed.
- A. V. Protchenko, A. D. Schwarz, M. P. Blake, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Angew. Chem., Int. Ed., 2013, 52, 568–571 CrossRef CAS PubMed.
- T. J. Hadlington, J. A. B. Abdalla, R. Tirfoin, S. Aldridge and C. Jones, Chem. Commun., 2016, 52, 1717–1720 RSC.
- S. Inoue and K. Leszczyńska, Angew. Chem., Int. Ed., 2012, 51, 8589–8593 CrossRef CAS PubMed.
- S. Yao, Y. Xiong and M. Driess, Organometallics, 2011, 30, 1748–1767 CrossRef CAS.
- A. Protchenko, P. Vasko, D. C. H. Do, J. Hicks, M. Á. Fuentes, C. Jones and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 1808–1812 CrossRef CAS PubMed.
- (a) W. Wang, S. Inoue, E. Irran and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 3691–3694 CrossRef CAS PubMed; (b) W. Wang, S. Inoue, S. Enthaler and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 6167–6171 CrossRef CAS PubMed.
- D. Gallego, S. Inoue, B. Blom and M. Driess, Organometallics, 2014, 33, 6885–6897 CrossRef CAS.
- (a) Y. Wang, A. Kostenko, S. Yao and M. Driess, J. Am. Chem. Soc., 2017, 139, 13499–13506 CrossRef CAS PubMed; (b) Y. Wang, A. Kostenko, T. J. Hadlington, M.-P. Luecke, S. Yao and M. Driess, J. Am. Chem. Soc., 2019, 141, 626–634 CrossRef CAS PubMed; (c) M.-P. Luecke, A. Kostenko, Y. Wang, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 12940–12944 CrossRef CAS PubMed.
- Y.-P. Zhou, S. Raoufmoghaddam, T. Szilvási and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 12868–12872 CrossRef CAS PubMed.
- H. F. Schaefer III, Acc. Chem. Res., 1982, 15, 283–290 CrossRef.
- See for examples: (a) M. Ma, A. Stasch and C. Jones, Chem. – Eur. J., 2012, 18, 10669–10676 CrossRef CAS PubMed; (b) W. Uhl, U. Schütz, W. Hiller and M. Heckel, Chem. Ber., 1994, 127, 1587–1592 CrossRef CAS; (c) Y. Xiong, S. Yao and M. Driess, Chem. – Eur. J., 2009, 15, 8542–8547 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details. CCDC 1963423 and 1963424. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc08680c |
| This journal is © The Royal Society of Chemistry 2020 |