
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal-free cyclobutene rearrangement in fused naphthalen-1-ones: controlled access to functionalized quinones†‡
Fernando
Herrera
a,
Amparo
Luna
 a,
Israel
Fernández
b and
Pedro
Almendros
*c
a,
Israel
Fernández
b and
Pedro
Almendros
*c
aGrupo de Lactamas y Heterociclos Bioactivos, Departamento de Química Orgánica, Unidad Asociada al CSIC, Facultad de Química, Universidad Complutense de Madrid, 28040-Madrid, Spain
bDepartamento de Química Orgánica I and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Facultad de CC. Químicas, Universidad Complutense de Madrid, 28040-Madrid, Spain
cInstituto de Química Orgánica General, IQOG-CSIC, Juan de la Cierva 3, 28006-Madrid, Spain. E-mail: palmendros@iqog.csic.es
First published on 19th December 2019
Abstract
The controlled synthesis of 1,4-naphthoquinones and tetraphene-7,12-diones, which bear the ABCD-ring of landomycins, has been accomplished directly through oxidative rearrangement of common stable precursors, namely, previously non-isolable cyclobuta[a]naphthalen-4(2H)-ones.
The ubiquity of the quinone moiety in natural products and organic materials justifies the continued interest in the synthesis of molecules containing this framework.1 The widespread occurrence of the cyclobutene nucleus in natural products and bioactive compounds, coupled to the use of this strained carbocycle as a building block in organic synthesis, triggered a renewed activity in the synthesis of cyclobutenes.2 However, the preparation of the quinone core from cyclobutenes has remained unexplored. Jiang and co-workers have recently communicated the cyclization reactions of allenynones toward naphthols using DABSO [DABCO·(SO2)2] and arenediazonium salts (Scheme 1a),3 or either in presence of alkynes (Scheme 1b),4 or β-ketonitriles (Scheme 1c),5 involving the generation of tricyclic cyclobutene intermediates. Aiming to extend the utility of allenynones, we planned to use allenyne precursors bearing substituents at the internal allene double bond. Worthy of note, the presence of the extra-substituent (R3 = Me, Ar) did allow for the isolation of previously unstable and non-isolable tricyclic cyclobutenes (R3 = H). Herein, we present a convenient method for the divergent synthesis of 1,4-naphthoquinones and tetraphene-7,12-diones through the oxidative reorganization of cyclobutene-fused naphthalen-1-ones (Scheme 1d).
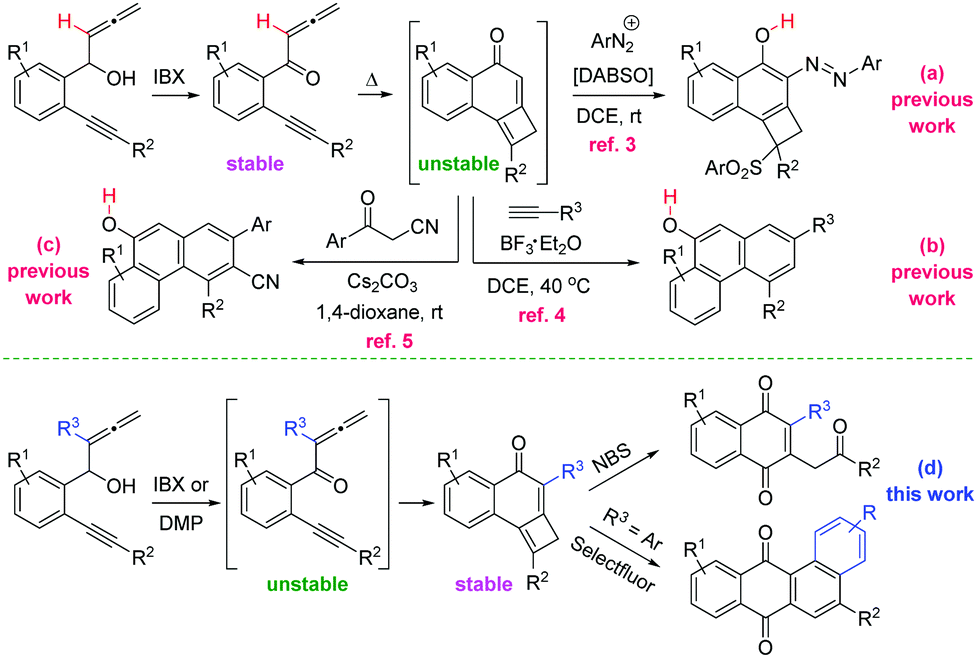 | ||
| Scheme 1 The allenynone framework as a platform for the synthesis of fused carbocycles: prior art and our method. | ||
Starting substrates, allenynols 2a–k, were prepared from alkynyl-benzaldehydes 1 and 3-substituted prop-2-ynyl bromides using an indium-mediated allenylation under Barbier conditions in aqueous media (Scheme 2). Further oxidation with IBX or Dess–Martin periodinane (DMP) to introduce the carbonyl moiety gives rise directly to cyclobutene-fused naphthalen-1-ones 3a–k (Scheme 2). It was found that the best results were systematically obtained in the presence of DMP. Because of the great reactivity imparted by the highly conjugated system in the initially formed non-isolable allenynones, [2+2] cycloaddition reactions take place spontaneously.6 Tricyclic cyclobutenes 3 were obtained with complete chemo- and regioselectivity in good yields.
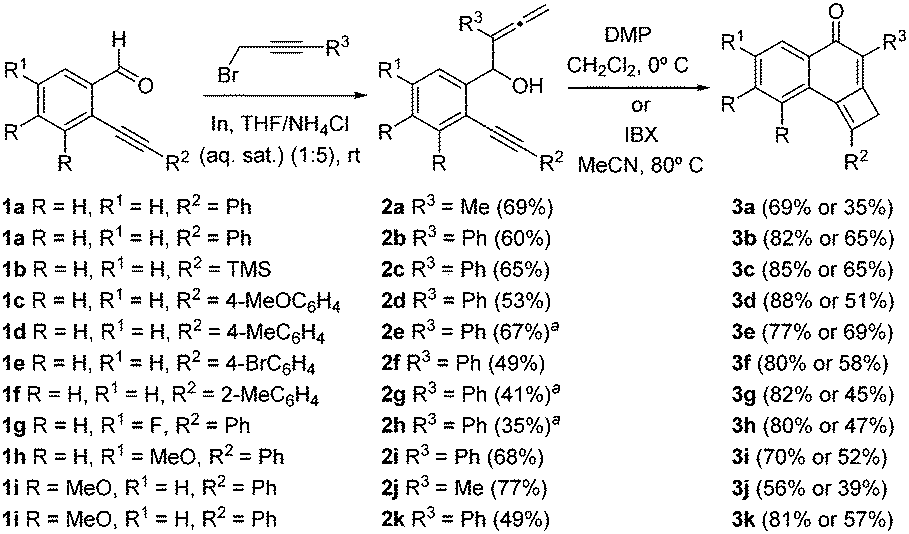 | ||
Scheme 2 Synthesis of tricyclic cyclobutenes 3a–k. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The reaction was carried out in THF/H2O (1:1). The reaction was carried out in THF/H2O (1:1). | ||
To explore the reactivity of cyclobutene-fused naphthalen-1-ones 3, at the beginning of the study we selected N-bromosuccinimide (NBS) as a convenient electrophilic source of bromine and tricyclic cyclobutene 3a as a model substrate. In the event, the use of THF as solvent led to reaction failure. Fortunately, the replacement of THF by acetonitrile provided 2-methyl-3-(2-oxo-2-phenylethyl)naphthalene-1,4-dione 4a,7 but only in a modest 29% yield. Because of the structure of 4a, it may be apparent that adventitious water in the reaction medium was necessary. To prove this assumption, we carried out the reaction with the addition of 5.0 equiv. of water. Indeed, in this way a significant improvement of the yield of 4a up to 72% was achieved (Scheme 3). With the optimal conditions in hand, we evaluated the influence of substituents at the different positions of tricycles 3. A variety of aromatic moieties were well tolerated both at R2 and R3, while an aliphatic substituent was accommodated at R3 (Scheme 3). Even precursor 3j bearing two electron-donating substituents (R = MeO) on the aromatic ring, conveniently afforded naphthalene-1,4-dione 4j after NBS treatment. By contrast, the reaction of TMS-cyclobutene 2c was not satisfactory. Interestingly, the presence of a fluorine atom at the benzene ring such as in tricyclic cyclobutene 3h resulted in an extra-bromination on the final adduct 4h-Br (Scheme 3). The same phenomenon was observed starting from tricycle 3a under otherwise identical reaction conditions but through mild heating, which resulted in the formation of 4a-Br (Scheme 3). Pleasingly, it has been reported that several 2-methyl-3-alkyl-naphthalene-1,4-diones related to bicycles 4 possess interesting biological activities.8
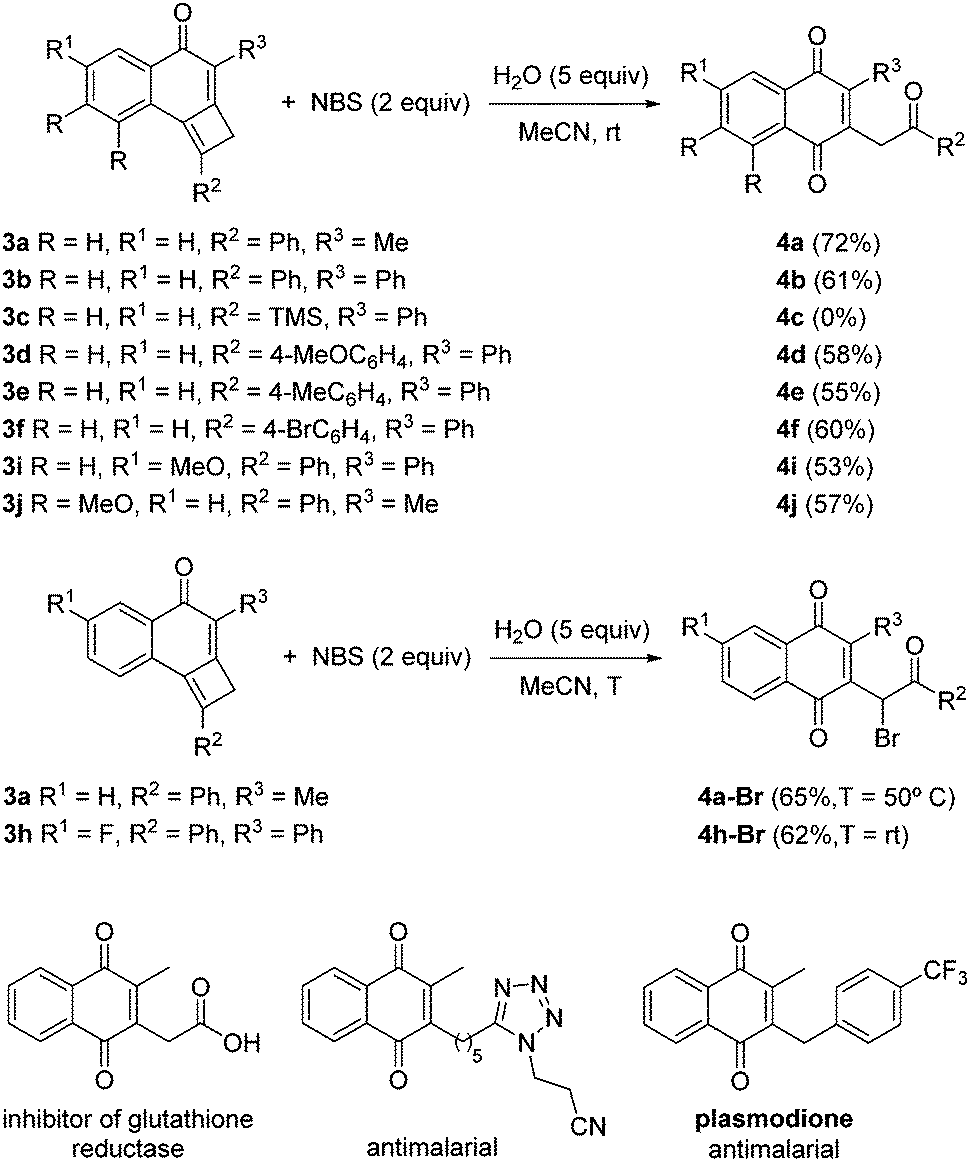 | ||
| Scheme 3 Synthesis of 2-substituted-3-(2-oxo-2-arylethyl)naphthalene-1,4-diones 4 and 4-Br. | ||
A plausible mechanism for the NBS-promoted genesis of 1,4-naphthoquinones 4 is delineated in Scheme 4. The reaction is presumed to possess a radical nature, which was based on the suspension of the transformation after the addition of TEMPO to the reaction medium. Initially, the formation of allylic radical species INT-1 should occur by bromine radical attack. The formation of this radical should be followed by oxidation to the carbocationic species INT-2 by single electron transfer (SET) to the succinimide. Next, water attack takes place with formation of bromohydrin INT-3 which is followed by HBr release. Hydration of the resulting intermediate cyclobutenol INT-4 leads to diol INT-5, which evolves into zwitterionic intermediate INT-6. According to Density Functional Theory calculations (see ESI‡), this step proceeds with a low activation barrier of only 10.6 kcal mol−1 (for 3a). A subsequent proton transfer leads to dihydroquinone 4H in a highly exergonic transformation (ΔGR = −40.3 kcal mol−1, for 3a), which is finally oxidized to its quinone form 4.
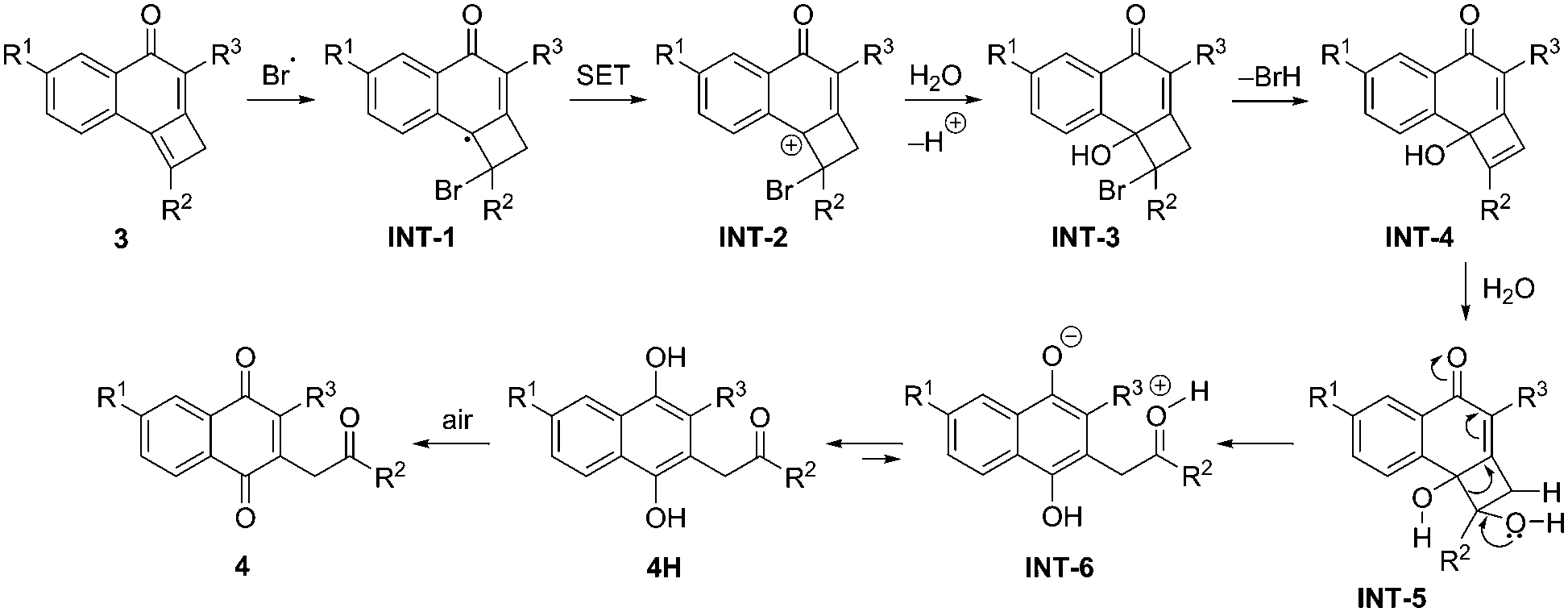 | ||
| Scheme 4 Mechanistic explanation for the NBS-promoted synthesis of 2-oxo-naphthalene-1,4-diones 4. | ||
Aiming to explore the effect of different reagents in the selective oxidative reorganization of cyclobutene-fused naphthalen-1-ones, we decided to expose tricycles 3 to the action of Selectfluor. The reaction of adduct 3a with Selectfluor was problematic and a complex mixture was obtained. By contrast, an encouraging result was obtained when adduct 3b having a phenyl group was used, because we unexpectedly isolated in a 19% yield tetracycle 5b that should arise from an angular benzannulation process. The presence of sodium bicarbonate in the reaction between Selectfluor and 3b caused an appreciable rise in the yield of 5b (Scheme 5). On this point, we next tested the scope of this rearrangement reaction using diverse fused-cyclobutenes 3 which contain a phenyl moiety at the cyclohexenone ring. A variety of differently substituted precursors 3, including fluoro- and methoxy-derivatives 3h and 3i were suitably rearranged. By contrast, the reaction of dimethoxy-substituted precursor 3k with Selectfluor was troublesome and the desired tetracycle 5k was not isolated in reasonable purity. Synthetically useful yields of tetracycles 5b and 5d–i were attained (Scheme 5). For all examples included in Scheme 5 no products of type 4 were observed. Noteworthy, tetracycles 5 bear the ABCD-ring of landomycins. Landomycins such as tetrangulol are a class of natural quinones characterized by an angular conjugated tetracyclic core, which confers them with interesting bioactivities.9 The major difficulty associated with the synthesis of landomycins is the formation of the B-ring due to hindrance issues. This elusive aspect is easily addressed in our preparation of the landomycin core.
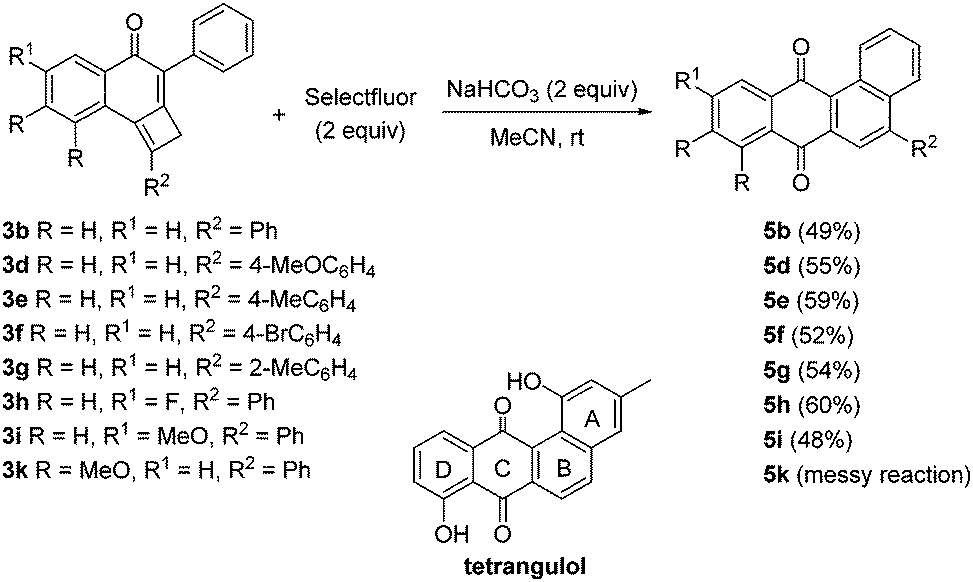 | ||
| Scheme 5 Synthesis of 5-aryltetraphene-7,12-diones 5b and 5d–i. | ||
In order to gain some mechanistic insights, control experiments were planned (Scheme 6). First, when tricycle 3b was treated with Selectfluor under the optimized conditions but using anhydrous acetonitrile, the yield of 5b decreased to 8% (Scheme 6). This result unveiled the origin of the oxygen at the C-ring, that should come from ambient water. Succeeding in performing a divergent preparation of 2-substituted-3-(2-oxo-2-arylethyl)naphthalene-1,4-diones 4 and 5-aryltetraphene-7,12-diones 5, we speculated about the possible intermediacy of 1,4-naphthoquinones 4 in the formation of angular tetracycles 5 in the presence of Selectfluor. When 1,4-naphthoquinone 4b was treated with Selectfluor and sodium bicarbonate under the optimized reaction conditions for the formation of pentacycles 5, the reaction failed and starting material 4b was fully recovered (Scheme 6). From the above experiment, it may be inferred that naphthalene-1,4-diones of type 4 should be discarded as intermediates for this reaction. The radical scavenger TEMPO effectively suppressed the formation of the required product, pointing to a radical reaction mechanism.
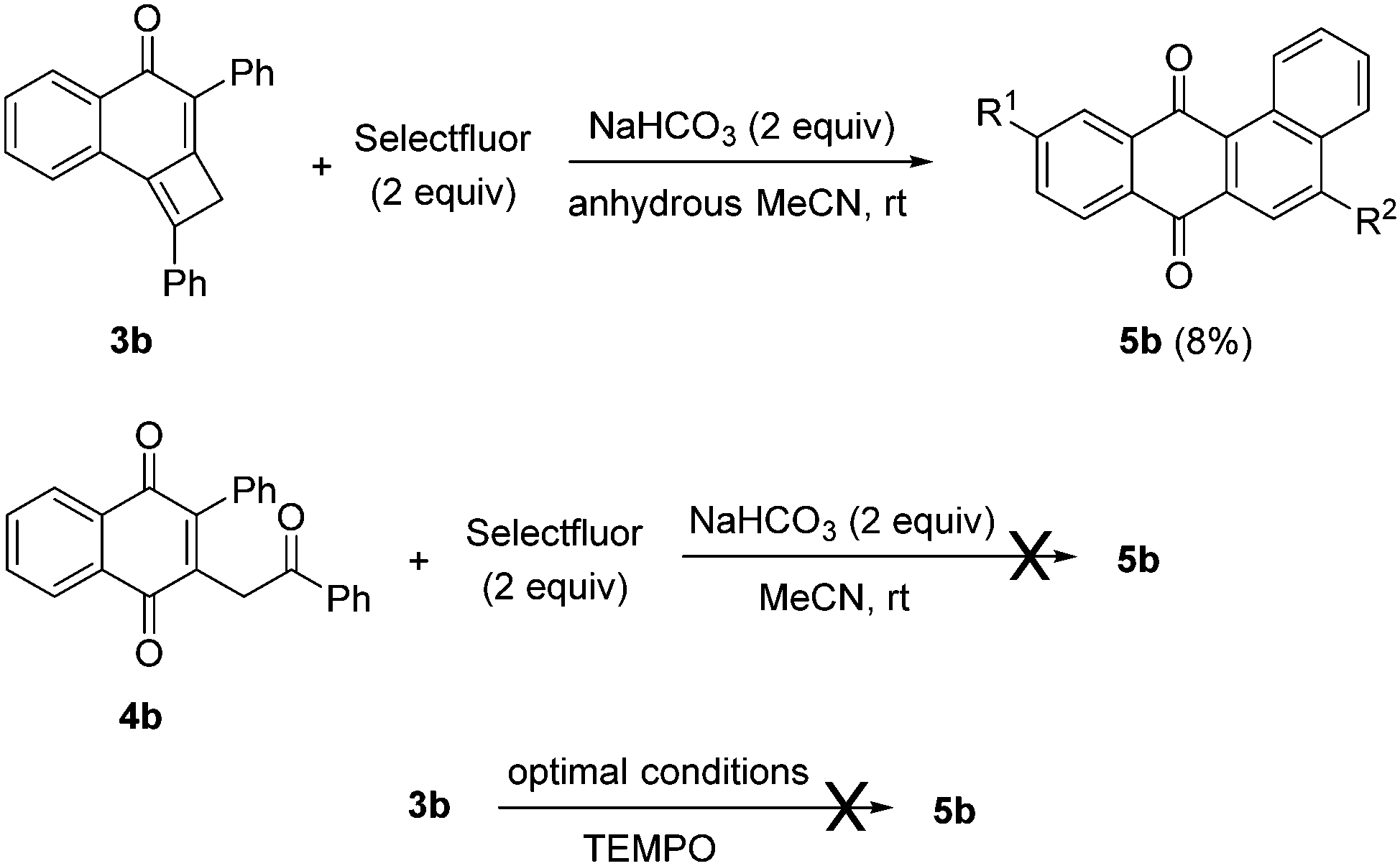 | ||
| Scheme 6 Control experiments. | ||
Based on the above experiments, we assume a mechanism as presented in Scheme 7 for the reaction of aryl-substituted cyclobutene-fused naphthalen-1-ones 3 with Selectfluor. Similar to the above-described reaction involving NBS, tricycles 3 can be converted into cationic intermediates INT-8 after Selectfluor treatment with concomitant formation of III. Next, this intermediate should suffer water attack to produce halohydrin INT-9, which evolves through cyclobutane ring opening followed by base-assisted HF release10 into the quinone INT-11. The so-formed polyene is able to undergo a 6π-electrocyclic ring closure to produce INT-12,11 which rapidly evolves into INT-13via a 1,3-hydrogen migration. According to DFT calculations on 3b (see ESI‡), this step proceeds with an activation barrier of 21.9 kcal mol−1 in a highly exergonic transformation (ΔGR = −28.2 kcal mol−1) driven by the gain in aromaticity in the system. Finally, INT-13 is oxidized into the observed tetracycles 5. Our calculations indicate that the concerted H2 release from either INT-13 or even from INT-12 thus directly forming 5 is unfeasible (ΔG‡ = 89.9 and 77.4 kcal mol−1, respectively). This points to a different mechanism for this final aromatization step,12 which is not evident to us at the moment.
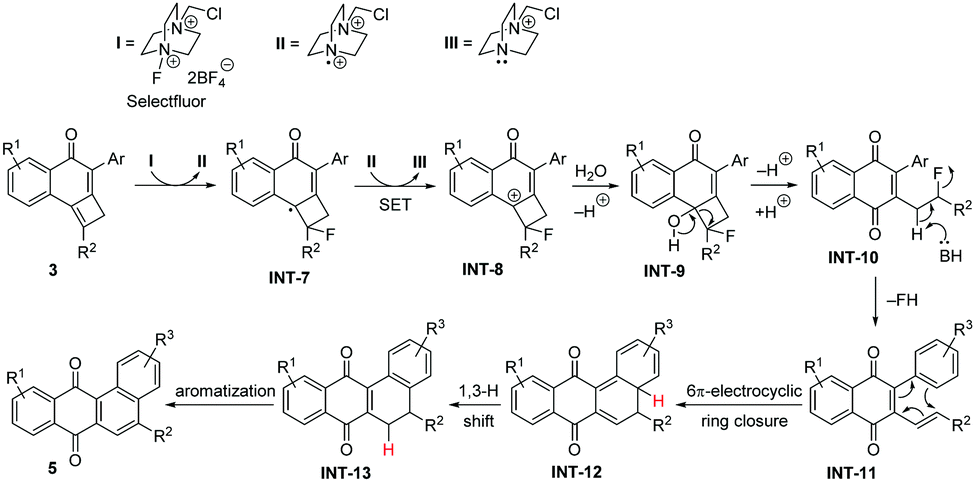 | ||
| Scheme 7 Mechanistic explanation for the Selectfluor-promoted synthesis of tetraphene-7,12-diones 5. | ||
In conclusion, we have developed a divergent outcome transformation of the previously non-isolable cyclobuta[a]naphthalen-4(2H)-one system to afford either 1,4-naphthoquinones or tetraphene-7,12-diones, which has been accomplished through the reorganization of the above fused tricyclic cyclobutenes in the presence of NBS or Selectfluor.
This work was supported in part by AEI (MICIU) and FEDER (Project PGC2018-095025-B-I00 to P. A. and CTQ2016-78205-P and CTQ2016-81797-REDC to I. F.). F. H. thanks UCM for a predoctoral contract. We are grateful to M. Tiemblo in our group for the preparation of compounds 2j, 2k and 3j and Prof. B. Alcaide for continued support.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- (a) S. Liu, T. Shen, Z. Luo and Z.-Q. Liu, Chem. Commun., 2019, 55, 4027 RSC and references therein. For representative reviews, see: ; (b) Y. Wang, S. Zhu and L.-H. Zou, Eur. J. Org. Chem., 2019, 2179 CrossRef CAS; (c) Y. Ando and K. Suzuki, Chem. – Eur. J., 2018, 24, 15955 CrossRef CAS PubMed; (d) E. J. Son, J. H. Kim, K. Kim and C. B. Park, J. Mater. Chem. A, 2016, 4, 11179 RSC; (e) S. N. Sunassee and M. T. Davies-Coleman, Nat. Prod. Rep., 2012, 29, 513 RSC; (f) C. Asche, Mini-Rev. Med. Chem., 2005, 5, 449 CrossRef CAS PubMed; (g) R. H. Thomson, Naturally Occurring Quinones IV, Blackie Academic & Professional, London, 1997 Search PubMed; (h) P. J. O’Brien, Chem.-Biol. Interact., 1991, 80, 1 CrossRef; (i) The Chemistry of the Quinonoid Compounds, ed. S. Patai and Z. Rappoport, Wiley, New York, 1988 Search PubMed.
- For recent references, see: (a) Q. Qin, X. Luo, J. Wei, Y. Zhu, X. Wen, S. Song and N. Jiao, Angew. Chem., Int. Ed., 2019, 58, 4376 CrossRef CAS PubMed; (b) G.-Q. Xu, J.-T. Xu, Z.-T. Feng, H. Liang, Z.-Y. Wang, Y. Qin and P.-F. Xu, Angew. Chem., Int. Ed., 2018, 57, 5110 CrossRef CAS PubMed; (c) T. Iwai, M. Ueno, H. Okochi and M. Sawamura, Adv. Synth. Catal., 2018, 360, 670 CrossRef CAS; (d) M. E. de Orbe, L. Amenós, M. S. Kirillova, Y. Wang, V. López-Carrillo, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2017, 139, 10302 CrossRef CAS PubMed; (e) M. Eisold, A. N. Baumann, G. M. Kiefl, S. T. Emmerling and D. Didier, Chem. – Eur. J., 2017, 23, 1634 CrossRef CAS PubMed; (f) B. Alcaide, P. Almendros and C. Lázaro-Milla, Chem. – Eur. J., 2016, 22, 8998 CrossRef CAS PubMed; (g) Y. Qiu, B. Yang, C. Zhu and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2016, 55, 6520 CrossRef CAS PubMed; (h) B. Alcaide, P. Almendros, I. Fernández and C. Lázaro-Milla, Chem. Commun., 2015, 51, 3395 RSC.
- (a) F. Liu, J.-Y. Wang, P. Zhou, G. Li, W.-J. Hao, S.-J. Tu and B. Jiang, Angew. Chem., Int. Ed., 2017, 56, 15570 CrossRef CAS PubMed . Fan has described the preparation of cyclobutanol-fused 2-nitro-naphthalen-1-ols: ; (b) T. Feng, Y. He, X. Zhang and X. Fan, Adv. Synth. Catal., 2019, 361, 1271 CrossRef CAS.
- (a) H. Li, W.-J. Hao, M. Wang, X. Qin, S.-J. Tu, P. Zhou, G. Li, J. Wang and B. Jiang, Org. Lett., 2018, 57, 4362 CrossRef PubMed; (b) R. Fu, M.-F. Li, P. Zhou, W.-J. Hao, S.-J. Tu and B. Jiang, Adv. Synth. Catal., 2019, 361, 2280 CrossRef CAS; (c) B.-Z. Tang, J.-Z. Li, A.-W. Zhang, W.-J. Hao, S.-J. Tu and B. Jiang, Adv. Synth. Catal., 2019, 361, 3394 CrossRef CAS.
- (a) H.-K. Sha, T. Xu, F. Liu, B.-Z. Tang, W.-J. Hao, S.-J. Tu and B. Jiang, Chem. Commun., 2018, 54, 10415 RSC; (b) J.-Y. Wang, F.-L. Xie, J.-Q. Hu, S.-Z. Yang, Y.-J. Wang, W.-J. Hao, S.-J. Tu and B. Jiang, Org. Biomol. Chem., 2018, 16, 7104 RSC.
- For a review on [2+2] cycloaddition chemistry with allenes, see: B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2010, 39, 783 RSC.
- Naphthoquinones have attracted considerable interest because of their relevant chemical and biological properties: (a) K. W. Wellington, RSC Adv., 2015, 5, 20309 RSC; (b) R. L. de Carvalho, G. A. M. Jardim, A. C. C. Santos, M. H. Araujo, W. X. C. Oliveira, A. C. S. Bombaça, R. F. S. Menna-Barreto, E. Gopi, E. Gravel, E. Doris and E. N. da Silva, Jr., Chem. – Eur. J., 2018, 24, 15227 CrossRef CAS PubMed and references therein; (c) A. S. Kumar, G. Thirupathi, G. S. Reddy and D. B. Ramachary, Chem. – Eur. J., 2019, 25, 1177 CrossRef CAS PubMed; (d) S. Peraka, M. A. Pasha, G. Thirupathi and D. B. Ramachary, Chem. – Eur. J., 2019, 25, 14036 CrossRef CAS PubMed.
- (a) C. Biot, H. Bauer, R. H. Schirmer and E. Davioud-Charvet, J. Med. Chem., 2004, 47, 5972 CrossRef CAS PubMed; (b) E. Davioud-Charvet, S. Delarue, C. Biot, B. Schwöbel, C. C. Böhme, A. Müssigbrodt, L. Maes, C. Sergheraert, P. Grellier, R. H. Schirmer and K. Becker, J. Med. Chem., 2001, 44, 4268 CrossRef CAS PubMed; (c) L. Feng, D. A. Lanfranchi, L. Cotos, E. Cesar-Rodo, K. Ehrhardt, A. A. Goetz, H. Zimmermann, F. Fenaille, S. A. Blandin and E. Davioud-Charvet, Org. Biomol. Chem., 2018, 16, 2647 RSC.
- C.-J. Sie, V. Patteti, Y.-R. Yang and K.-K. T. Mong, Chem. Commun., 2018, 54, 1885 RSC and references therein.
- (a) L. Xu, Q. Zhang, Q. Xie, B. Huang, J.-J. Dai, J. Xu and H.-J. Xu, Chem. Commun., 2018, 54, 4406 RSC; (b) C. Gerleve, M. Kischkewitz and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 2441 CrossRef CAS PubMed; (c) X.-Q. Chu, B.-Q. Cheng, Y.-W. Zhang, D. Ge, Z.-L. Shen and T.-P. Loh, Chem. Commun., 2018, 54, 2615 RSC; (d) S.-L. Zhang and J.-J. Dong, Org. Lett., 2019, 21, 6893 CrossRef CAS PubMed; (e) C. Li, J.-M. Yuan, W. Chen, Y. He, J. Huang, Y. Huang, Q. Xiao, J. Sheng and C. Huang, Chem. – Asian J., 2019, 14, 2584 CrossRef CAS PubMed; (f) J. Qian, J. Zhang, H. Yang, L. Kang and G. Jiang, Chem. Sci., 2019, 10, 8812 RSC; (g) Y. Chen, L. Li, X. He and Z. Li, ACS Catal., 2019, 9, 9098 CrossRef CAS.
- For recent use of 6π-electrocyclization in the synthesis of polycycles, see: (a) H. Kim, Y. J. Hwang, I. Han and J. M. Joo, Chem. Commun., 2018, 54, 6879 RSC; (b) B. Alcaide, P. Almendros, C. Lázaro-Milla and P. Delgado-Martínez, Chem. – Eur. J., 2018, 24, 8186 CrossRef CAS PubMed; (c) M. P. Ball-Jones, J. Tyler, H. Mora-Radó, W. Czechtizky, M. Méndez and J. P. A. Harrity, Org. Lett., 2019, 21, 6821 CrossRef CAS PubMed; (d) C. Elgindy, J. S. Ward and M. S. Sherburn, Angew. Chem., Int. Ed., 2019, 58, 14573 CrossRef CAS PubMed.
- For recent mechanistic proposals involving the conversion of intermediate dihydroarenes into the corresponding arenes which is associated with an oxidation step, probably due to the presence of atmospheric oxygen, see: (a) A. J. Ansari, A. A. Wani, A. K. Maurya, S. Verma, V. K. Agnihotri, A. Sharon, P. V. Bharatam and D. M. Sawant, Chem. Commun., 2019, 55, 14825 RSC; (b) G. Bianchini, P. Ribelles, D. Becerra, M. T. Ramos and J. C. Menéndez, Org. Chem. Front., 2016, 3, 412 RSC; (c) Q. Xu, P. Gu, F. Wang and M. Shi, Org. Chem. Front., 2015, 2, 1475 RSC.
Footnotes |
| † In memory of Prof. Odón Arjona. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data, and copies of NMR spectra for all new compounds. See DOI: 10.1039/c9cc08628e |
| This journal is © The Royal Society of Chemistry 2020 |