
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and application of aminoacridinium organophotoredox catalysts
Bouthayna
Zilate
,
Christian
Fischer
 and
Christof
Sparr
*
and
Christof
Sparr
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, CH-4056, Basel, Switzerland. E-mail: christof.sparr@unibas.ch
First published on 30th January 2020
Abstract
Recent developments in preparative photocatalysis have reshaped synthetic strategies and now represent an integral part of current organic chemistry. Due to their favourable electrochemical and photophysical properties, the nowadays most frequently used photocatalysts are based on precious Ru- and Ir-polypyridyl complexes. Apart from that, organic catalysts such as the acridinium salts are now commonly employed to complement transition metals to provide potentially sustainable strategies amenable to large-scale synthesis. In this feature article, the design, synthesis and application of aminoacridinium photoredox catalysts as well as their exceptionally broad range of redox properties are highlighted. Due to their modularity, this burgeoning class of organophotocatalysts is anticipated to contribute significantly to synthetic methodology development and the translation to a wide range of innovative implementations.
 Bouthayna Zilate | Bouthayna Zilate was born in 1988 in Casablanca (Morocco), studied pharmacy at the University of Paris V where she obtained her MSc with Prof. Hervé Galons working on inhibitors of cyclin-dependent kinases (CDK). In 2016, she started a PhD in the group of Prof. Sparr at the University of Basel. She's currently investigating novel approaches for the synthesis of acridinium salts and their application in photoredox catalysis. |
 Christian Fischer | Christian Fischer grew up in Bern and studied chemistry at the University of Basel. He worked with Prof. Oliver S. Wenger and Prof. Karl Gademann and completed his Master's thesis in the group of Prof. Christof Sparr. In his PhD studies he focused on the development of new, sustainable methods and catalysts. In December 2018, he joined the research group of Prof. Dirk Trauner (New York University) as an SNSF postdoctoral fellow, to investigate the photopharmacological control of biological processes. |
 Christof Sparr | Christof Sparr received his PhD from the ETH Zurich (Switzerland) working in the group of Prof. Ryan Gilmour. He then joined the groups of Prof. Dieter Seebach (ETH Zurich) and subsequently Prof. Steven V. Ley (University of Cambridge, UK). In 2013, Christof became habilitand mentored by Prof. Karl Gademann and since 2016, Christof has worked as Assistant Professor at the University of Basel. He is the recipient of the ETH silver medal, an SNSF starting grant, the Werner Prize of the Swiss Chemical Society 2017 and the Ruzicka Prize of the ETH Zurich 2018. |
1. Introduction
Polypyridyl transition metal systems have been transformative for the field of photoredox catalysis with ruthenium and iridium catalysts representing particularly versatile scaffolds (Scheme 1).1–4 The wide range of properties due to the tunability of their ligands make them exceptionally valuable catalysts for photochemical transformations. With the development of fully organic alternatives, the area opened up towards potentially sustainable processes.5–7 Among them, acridinium salts pioneered by Fukuzumi8,9 are known to have remarkable photophysical properties, complementary to those of polypyridyl transition metal structures. Their high reduction potential in the excited state (often E1/2 [P*/P−] > 2 V vs. SCE) and their robustness, pH independence, and high solubility in a variety of solvents, render them valuable photocatalysts for a wide range of preparative transformations.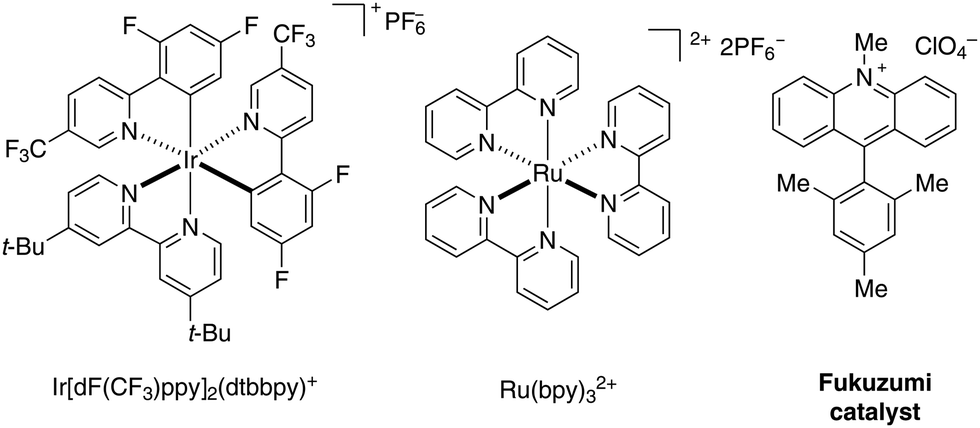 | ||
| Scheme 1 Selection of commonly used photoredox catalysts. | ||
2. Design and synthesis of acridinium salts
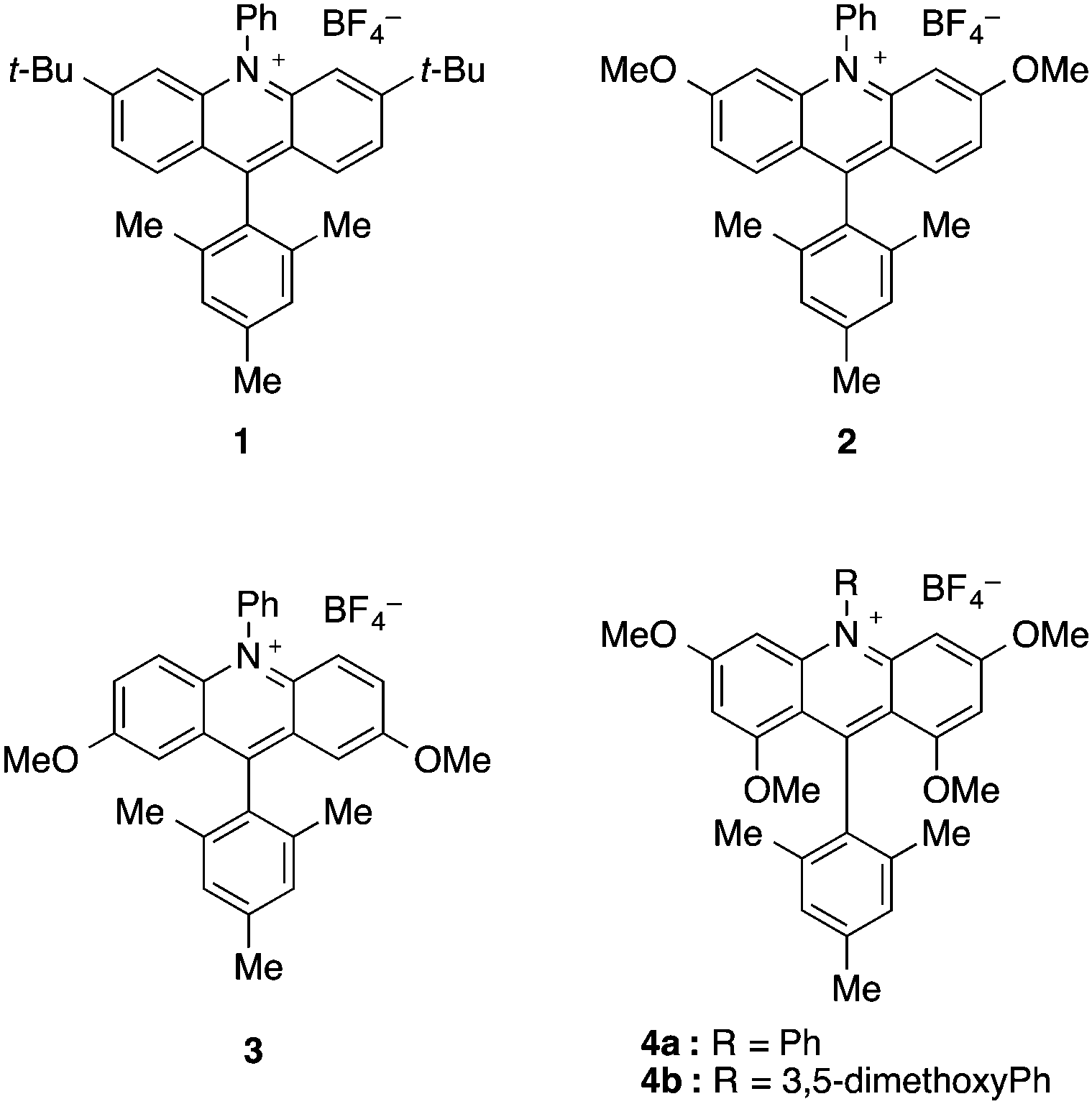
The design of acridinium dyes initially evolved towards an increased chemical stability. First, Fukuzumi integrated a bulky mesityl group at the C9 position to prevent nucleophilic addition that results in photobleaching.8 Moreover, the introduction of electron donating groups favours charge transfer by the stabilizing mesityl moiety.10 As a consequence, the Mes-Me-Acr+ displays unique oxidizing properties (E1/2 [P*/P−] = +2.08 V vs. SCE), with a suitable lifetime of the singlet and triplet excited state, which renders it a valuable catalyst for numerous photochemical reactions.11–14To further overcome competitive dealkylation processes, Nicewicz developed N-arylated acridinium salts with higher chemical stability and an extended range of redox properties (E1/2 [P*/P−] = 1.62–2.08 V vs. SCE, Scheme 2).15 A series of symmetrically substituted acridinium salts were prepared, with alkyl or methoxy substituents attached to the acridinium core.
 | ||
| Scheme 2 Representative photocatalysts reported by Nicewicz and co-workers. | ||
A one step synthesis featuring a triarylamine precursor in a Friedel–Crafts reaction with a benzoyl chloride derivative delivered the desired acridinium salt 6 with a tetrasubstituted core in good yields (71–81%, Scheme 3). The construction of other acridinium salts with a disubstituted core proved to be more challenging, requiring several steps including an addition of a Grignard reagent to an acridone. For a modular synthesis of acridinium salts (7, X = N) and other heterocyclic analogues such as xanthylium dyes (7, X = O), we thus investigated the use of 1,5-bifunctional organometallic reagents that allow the direct conversion of a broad range of carboxylic acid esters.16 Recently, Nicewicz and co-workers designed an efficient synthesis of acridinium photocatalysts starting from xanthylium salts.17 The rapid assembly of a xanthylium dye was thereby combined with a condensation reaction with an aniline to give the corresponding acridinium salt 1.
 | ||
| Scheme 3 Selected synthetic strategies for acridinium salts. | ||
Furthermore, an elegant strategy consisting of an oxidative Ugi-type reaction for the synthesis of a novel class of imide-acridinium salts has been developed by Mancheño and Alemán (Scheme 4).18 Starting from N-substituted acridanes, a copper-catalysed oxidation takes place at the C9 position to give a cationic intermediate that reacts with the isocyanide. After addition of the in situ formed benzoate, the intermediate is concomitantly rearranged in an Ugi-type fashion to produce the C9-substituted imide-acridanes 10. A subsequent aromatization by hydride abstraction carried out by tritylperchlorate delivered the desired acridinium salt 11. Photophysical and photoredox studies of these catalysts have been conducted.19 Direct applications in photoredox catalysis were subsequently demonstrated with a suitable activity in several benchmarking transformation including anti-Markovnikov hydrofunctionalization of alkenes and a dehydrogenative lactonization.
 | ||
| Scheme 4 Ugi reaction for the synthesis of acridinium photocatalysts and their application. | ||
3. Applications in photoredox chemistry
A rapidly growing number of chemical transformations employs acridinium salts as catalysts.20–23 Photooxidations, polar radical crossover cycloaddition reactions, C–H functionalization, cross-coupling and cross-dehydrogenative reactions, are merely selected recent examples of the use of acridinium salts in photoredox catalysis.24–30 A recent example from the Nicewicz group emphasises their unique features by the radical conjugate addition of N-carbamate-protected secondary amines (Scheme 5).22 Notably, the reduction potential required for the formation of the α-radical is too high for common Ru- or Ir- based photocatalysts (N-Boc-piperidine, Eox = 1.96 V vs. SCE). The application of strong excited-state oxidants such as the 9-mesityl-3,6-di-t-butyl-10-phenylacridinium salt or the Fukuzumi catalyst is hence required. This afforded the α-aminoalkylation of various carbamate protected secondary amines, including the substrate-controlled stereoselective synthesis of the natural product (+)-monomorine 15. | ||
| Scheme 5 Acridinium photocatalyzed α-alkylation of carbamate-protected secondary amines demonstrated by the synthesis of (+)-monomorine (15). | ||
Moreover, an acyl radical generation for the synthesis of asymmetric ketones was described by Fagnoni and co-workers (Scheme 6).31 Starting from readily prepared acylsilanes, the oxidation to the corresponding radical cation by Fukuzumi's catalyst led to the aliphatic acyl radical after loss of the TMS group. The addition to a Michael acceptor followed by reduction and protonation delivered a variety of asymmetric ketones.
 | ||
| Scheme 6 Photocatalytic acylation of Michael acceptors. | ||
Recently, Glorius and co-workers disclosed a carbonyl olefin cross-metathesis catalysed by an acridinium salt derived from Nicewicz's 3,6-di-tert-butyl acridinium catalyst (Scheme 7).32 1,3-Diols are thereby generated from aldehydes 20 and alkenes 19 upon visible-light irradiation that subsequently undergo acid-promoted Grob-type fragmentations. After the elimination of acetone, the cross-metathesis product is thereby formed.
 | ||
| Scheme 7 Carbonyl olefin cross-metathesis for the synthesis of disubstituted trans alkenes. | ||
Using a cascade of C–H and C–C bond cleavage steps, the group of Leonori achieved an intriguing remote functionalization of nitriles and ketones induced by the photoexcited Mes-Acr-Me+ catalyst (Scheme 8).33 The key step of this transformation lies in the formation of an iminyl radical arising from the SET oxidation and deprotonation of a carboxylic acid containing cyclic oxime 22. For instance, a stepwise sequence of an abnormal Beckmann fragmentation and a fluorination gives access to a broad range of distal fluorinated nitriles. This strategy could also be applied to the synthesis of γ-fluorinated and chlorinated ketones. After SET oxidation of a linear oxime 24, a 1,5-hydrogen abstraction provides a nucleophilic γ-radical that reacts with N-chlorosuccinimide or selectfluor to deliver the targeted compounds.
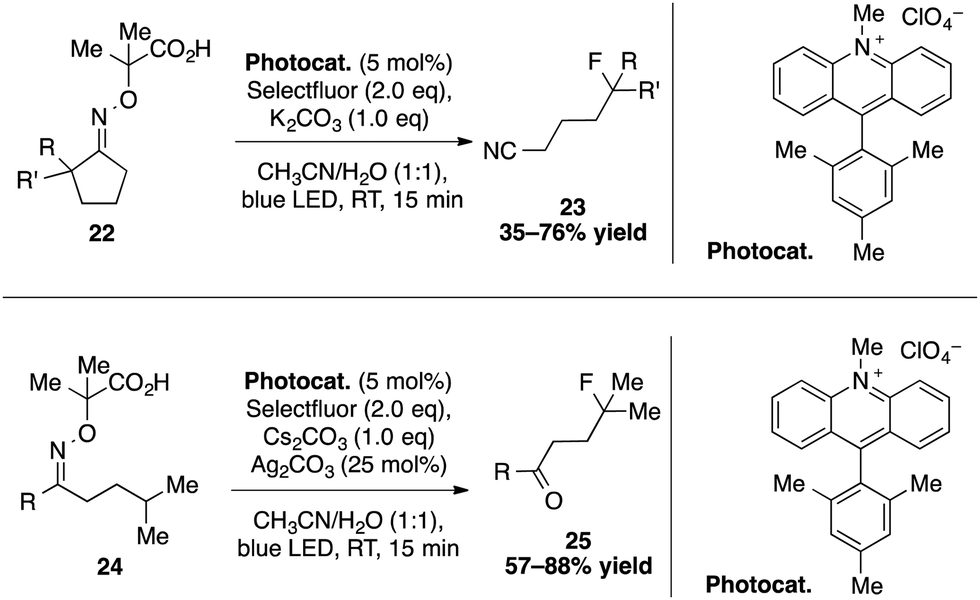 | ||
| Scheme 8 Remote fluorination and chlorination via iminyl radicals. | ||
A photocatalyzed three-component carbofluorination for the synthesis of α-fluoro-α-amino acid derivatives has been developed by Molander and co-workers (Scheme 9).34 Upon irradiation of the Mes-Acr-Me+ catalyst, an alkyltrifluoroborate reagent is oxidized to give an alkyl radical that adds to the dehydroalanine to generate an α-amino radical intermediate, which reacts with selectfluor to provide the α-fluoro-α-amino acids 27.
 | ||
| Scheme 9 Photocatalyzed synthesis of α-fluoro-α-amino acids. | ||
The Nicewicz group furthermore investigated an organophotoredox catalysed C–H fluorination of arenes with 18F (Scheme 10).35 Using the 9-mesityl-3,6-di-t-butyl-10-phenylacridinium salt as a photooxidant, TEMPO as a redox co-mediator and caesium fluoride combined with tetrabutylammonium bisulfate to generate TBAF in situ, the 18F-fluorination of a wide range of arenes, heterocycles and even bioactive compounds was enabled. Their approach was expanded to radiofluorinations for PET imaging applications in diagnosis and pharmacological studies.
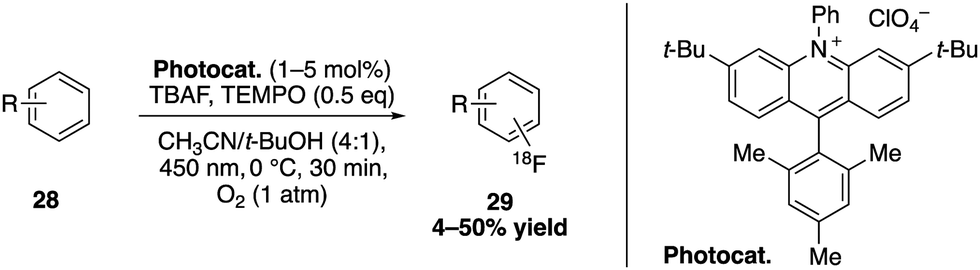 | ||
| Scheme 10 Direct 18F-fluorination catalysed by acridinium salts. | ||
Several direct photoredox catalysed C(sp2)–H functionalization reactions induced by a dual catalytic cobalt/Mes-Acr-Me+ combination were explored by the group of Lei.36,37 Among them an oxidative phosphonylation of arenes for the synthesis of valuable molecular scaffold is described (Scheme 11).38 The key step of this reaction is the SET oxidation of the arene to give a radical cation that reacts with the trialkylated phosphite. The resulting intermediate is then oxidized to a cationic intermediate which is subsequently deprotonated. By loss of an alkyl moiety, a wide range of aryl phosphonate products were obtained, even allowing the late stage functionalization of druglike compounds.
 | ||
| Scheme 11 Photooxidative phosphonylation of arenes. | ||
Extending the application of their dual catalytic cobalt/acridinium salt process, the same group also performed a photooxidative [4+2] annulation between aromatic ketimine derivatives and styrenes (Scheme 12).39,40 The SET oxidation of the alkene substrate by the excited Mes-Acr-Me+ catalyst thereby produces a radical cation that reacts with the nucleophilic aromatic ketimine to give a stable benzylic radical intermediate. A subsequent sequence consisting of a cyclization, oxidation and deprotonation allows to obtain highly substituted 3,4-dihydroisoquinolines with excellent trans diastereoselectivity.
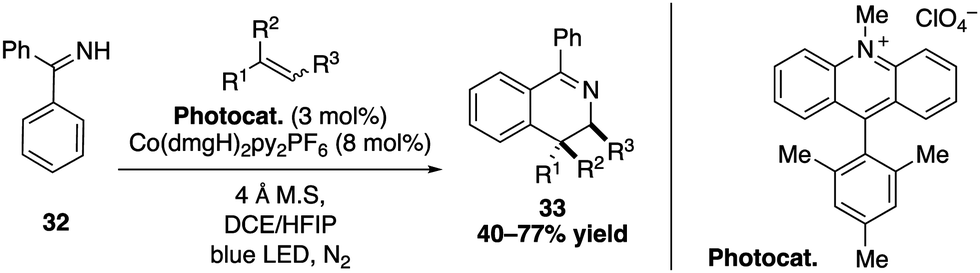 | ||
| Scheme 12 Photooxidative [4+2] annulation for the synthesis of 3,4-dihydroisoquinolines derivatives. | ||
The group of Kanai investigated a different type of dual catalytic palladium/acridinium systems for the catalytic dehydrogenation of N-heterocycles under visible light irradiation at ambient temperature (Scheme 13).41 For tetrahydronaphthalenes, a thiophosphoric imide organocatalyst was added as a third component, allowing in this catalytic process the formation of hydrogen gas under mild conditions.
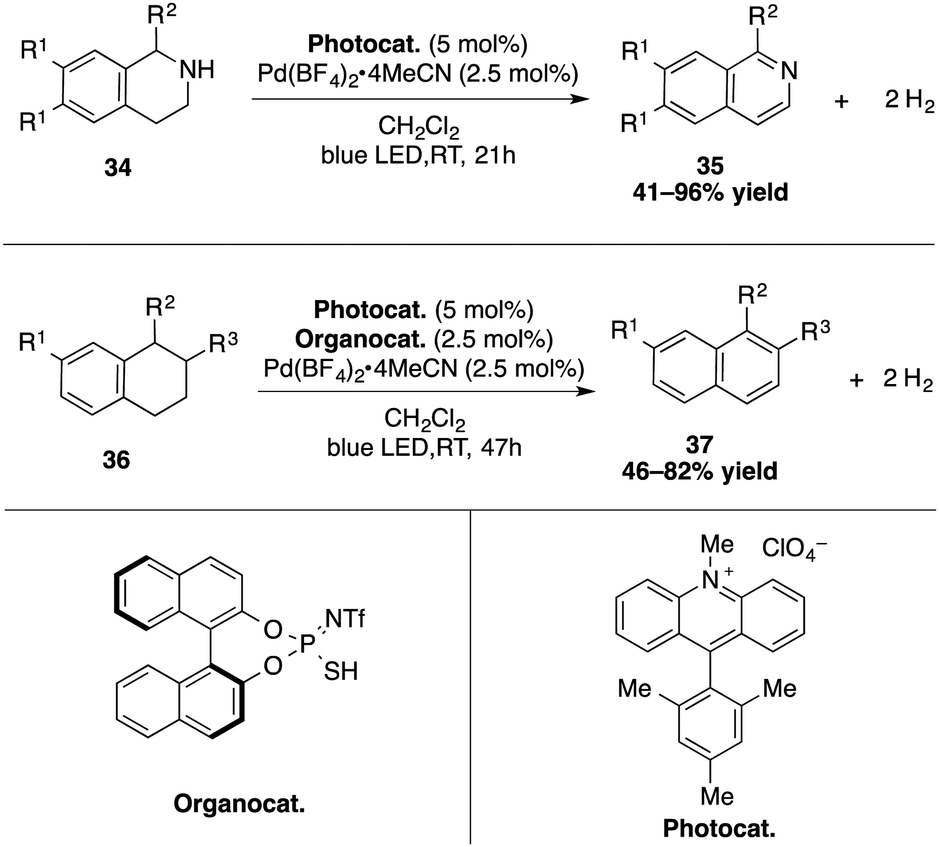 | ||
| Scheme 13 Catalytic dehydrogenation of N-heterocycles and tetrahydronaphthalene derivatives. | ||
Exploiting the properties of the Mes-Acr-Me+, Rueping and co-workers reported a cross-coupling of allylic C(sp3)–H bonds with aryl- and vinylbromides, using a combination of nickel and an acridinium photocatalyst (Scheme 14).16,42 After the oxidative addition of aryl- or vinylbromide to the nickel, a triplet–triplet energy transfer was proposed to take place between the excited acridinium salt and the nickel species. Cleavage of the halogen-metal-bond then generates the radical bromide, which abstracts a hydrogen from the alkene to form an allylic radical compound. The reaction with the Ni intermediate then undergoes a reductive elimination to release the allylarene 39 or corresponding 1,4-dienes.
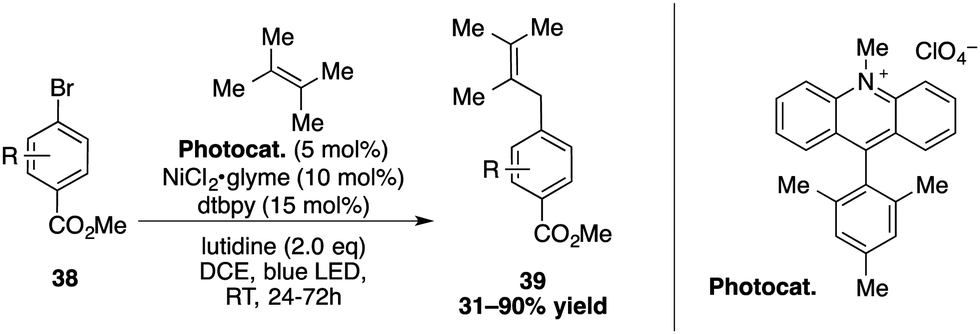 | ||
| Scheme 14 Nickel dual catalytic cross-coupling for allylic arylation. | ||
Acridinium salts are not limited to visible light-induced catalysis but can also be used as organocatalyst based on their ground state redox properties. Taking advantage of those properties, Ooi and co-workers designed an interesting acridinium betaine catalyst that enables PCET (proton-coupled electron transfer) and applied it to the oxidative dimerization of oxindoles under air (Scheme 15).43 The 9-mesityl acridinium moiety acts as the redox active unit whereas the phenoxide represents the substrate activation site. Using this catalyst, a successful homodimerization of 3-aryl oxindoles could be achieved.
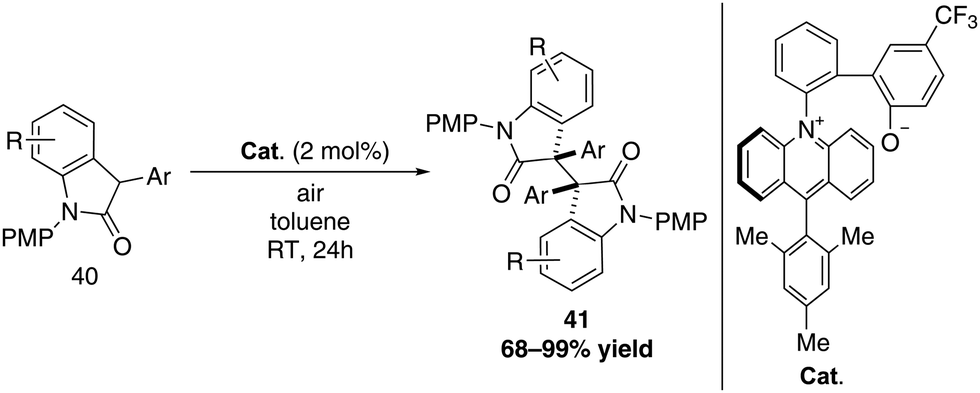 | ||
| Scheme 15 Dimerization of 3-aryl oxindoles catalysed by an acridinium betaine. | ||
4. Modulation of aminoacridinium photocatalysts
The evolution of the synthesis of acridinium salts towards more modular approaches allowed the diversification of their scaffold in order to tune their photophysical properties. To further modulate this class of catalysts, the authors aimed at the efficient synthetic preparation of aminoacridinium salts, which allow the lowering of the redox potential of the Fukuzumi catalyst (E1/2 [P*/P−] = +2.06 V vs. SCE) up to the range of the Ir-based photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 (E1/2 [P*/P−] = +1.21 V vs. SCE, Table 1).16 With 1,8-substituted and asymmetric acridinium salts, a wide range of excited state reduction potential ranging from +1.19 V to +1.88 V against SCE was reached.44,45 Also acridinium salts without amino functionalities were accessible, providing very high excited state reduction potentials of up to +2.32 V against SCE.46 The excited state lifetimes of the photocatalysts are typically in the order of nanoseconds which render them suitable for photoredox catalysis.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Dye | λ abs,max [nm] | λ em,max [nm] | E 0,0 [eV] | E 1/2(P/P−) [V] | E 1/2(P*/P−) [V] | τ [ns] | HOMO–LUMO transition |
| Fukuzumi catalyst | 425 | — | 2.57 | −0.57 | +2.08 | 6 | CT |
| 1 | 420 | 517 | — | 0.59 | +2.08 | 14.4 | — |
| 2 | 407 | 525 | — | −0.71 | +2.01 | 3.0 | — |
| 3 | 466 | 545 | — | −0.57 | +1.90 | 18.7 | — |
| 4a | 412 | 550 | — | −0.84 | +1.62 | 1.3, 8.9 | — |
| 4b | 414 | 550 | — | −0.82 | +1.65 | 1.3, 12.3 | — |
| 42 | 501 | 531 | 2.41 | −1.15 | +1.26 | — | — |
| 43 | 502 | 534 | 2.40 | −1.10 | +1.30 | — | — |
| 44 | 506 | 534 | 2.39 | −1.13 | +1.26 | — | — |
| 45 | 506 | 532 | 2.39 | −1.14 | +1.25 | — | — |
| 46 | 503 | 534 | 2.40 | −1.12 | +1.28 | — | — |
| 47 | 503 | 534 | 2.40 | −1.15 | +1.25 | 2.2 | π–π* |
| 48 | 504 | 533 | 2.40 | −1.15 | +1.25 | — | — |
| 49 | 438 | 499 | 2.77 | −0.56 | +2.21 | — | — |
| 50 | 426 | 512 | 2.83 | −0.51 | +2.32 | — | — |
| 51 | 498 | 540 | 2.40 | −1.19 | +1.21 | 4.4 | — |
| 52 | 503 | 595 | 2.23 | −0.47 | +1.76 | 3.1 | — |
| 53 | 501 | 584 | 2.25 | −0.94 | +1.31 | 4.7 | — |
| 54 | 494 | 567 | 2.30 | −0.62 | +1.68 | 5.9 | — |
| 55 | 497 | 531 | 2.39 | −0.51 | +1.88 | 4.1 | — |
| 56 | 497 | 579 | 2.31 | −0.51 | +1.80 | 3.0 | — |
| 57 | 497 | 576 | 2.33 | −0.52 | +1.81 | 2.7 | — |
| 58 | 506 | 575 | 2.30 | −0.83 | +1.47 | 0.9, 4.4 | π–π* |
| 59 | 480 | 634 | 2.25 | −0.56 | +1.69 | 1.2, 3.3, 16.8 | Mixed |
| 60 | 514 | 574 | 2.29 | −0.90 | +1.39 | 1.1, 7.2 | π–π* |
| 61 | 516 | 578 | 2.27 | −0.87 | +1.40 | 1.0, 6.2 | Mixed |
| 62 | 473 | 635 | 2.29 | −0.48 | +1.81 | 1.0, 3.0, 17.3 | π–π* |
| 63 | 480 | 635 | 2.26 | −0.53 | +1.73 | 1.0, 4.5 | CT |
| 64 | 479 | 637 | 2.23 | −0.54 | +1.69 | 1.0, 9.9 | π–π* |
| 65 | 511 | 576 | 2.29 | −0.89 | +1.40 | 1.0, 6.9 | π–π* |
| 66 | 583 | 723 | 1.94 | −0.71 | +1.23 | 1.5, 5.3 | π–π* |
| 67 | 479 | 632 | 2.25 | −0.57 | +1.68 | 1.4, 12.1 | Mixed |
| 68 | 513 | 573 | 2.29 | −0.89 | +1.40 | 1.1, 6.8 | π–π* |
| 69 | 590 | 755 | 1.87 | −0.68 | +1.19 | 0.9, 5.0 | π–π* |
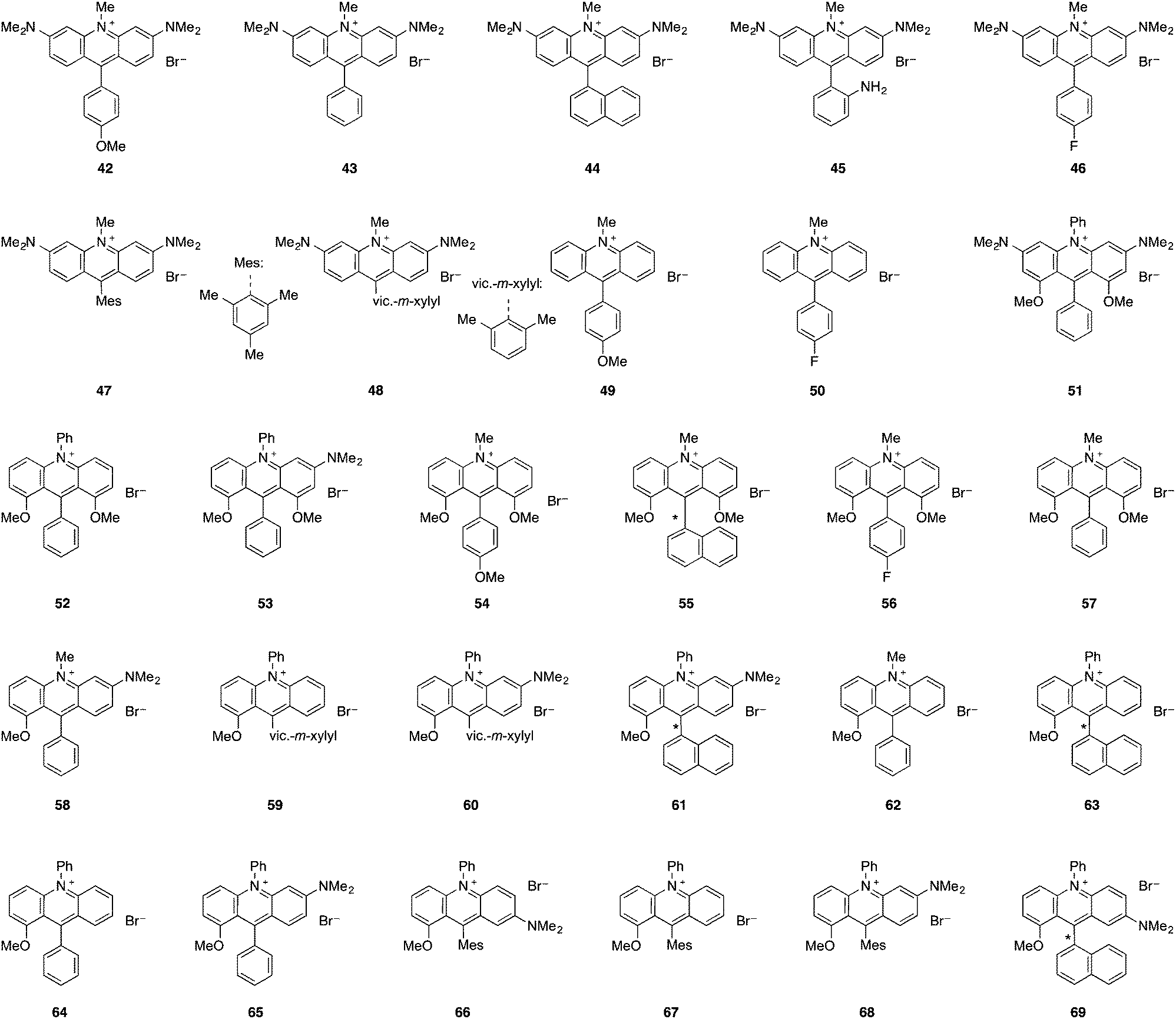
5. Modular synthesis of acridinium salts
In order to access this range of acridinium salts, individual strategies were employed for a modular synthetic preparation. For the synthesis of symmetrically or unsubstituted acridinium salts, such as diamino derivatives, a 1,5-bifunctional organomagnesium reagent was added to a variety of carboxylic acid esters to provide a broad range of acridinium derivatives, some of which were scaled up to five grams (Scheme 16).16,46 | ||
| Scheme 16 1,5-bifunctional organomagnesium reagent for the synthesis of unfunctionalized and diamino acridinium dyes. | ||
The synthesis of 1,8-dimethoxy substituted acridinium dyes was achieved through a double directed ortho-metalation (DoM) to generate the 1,5-organodilithium reagent that was engaged in a direct ester to heterocycle transformation with variation of the carboxylic acid esters (Scheme 17).
 | ||
| Scheme 17 Double directed ortho-metalation (dDoM) for the synthesis of 1,8-dimethoxy substituted acridinium salts. | ||
This approach also enabled the synthesis of atropisomeric acridinium salts with a 1,8-dimethoxy-peri-substitution and 9-naphthyl moiety (72, Scheme 18).46 After reduction of the racemic acridinium, the enantiomers of the leuco-form with a diastereomeric ratio of 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 were separated by HPLC and reoxidized by chloranil to give the two atropisomerically pure compounds. These compounds display a remarkable configurational stability with bond-rotational barriers of ΔG‡298K = 124 kJ mol−1 and ΔG‡298K = 127 kJ mol−1 respectively.
:3 were separated by HPLC and reoxidized by chloranil to give the two atropisomerically pure compounds. These compounds display a remarkable configurational stability with bond-rotational barriers of ΔG‡298K = 124 kJ mol−1 and ΔG‡298K = 127 kJ mol−1 respectively.
 | ||
| Scheme 18 Synthesis of atropisomeric acridinium salts. | ||
An entirely modular synthetic strategy for asymmetric acridinium salts was envisaged based on a combination of directed ortho-metalation (DoM) and halogen metal exchange (X–M) processes (Scheme 19). With this strategy, a broad panel of acridinium salts was obtained with various substitution patterns.45
 | ||
| Scheme 19 Modular synthesis of asymmetric acridinium salts. | ||
6. Catalytic performance
The performances of these photocatalysts was verified in several benchmarking reactions. A photoredox/nickel dual catalytic decarboxylative cross coupling of Boc-L-proline (75) with methyl 4-iodobenzoate (76), originally reported by Doyle and MacMillan with an iridium photocatalyst, was the starting point of our inquiry.47 The C–C coupling product 77 was obtained in very good yield (86%) using the mesityl-substituted diaminoacridinium catalyst 47 (Scheme 20).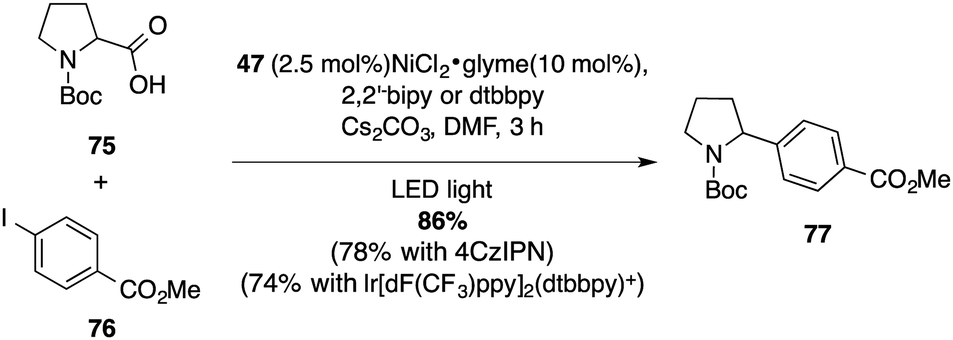 | ||
| Scheme 20 Photoredox/Ni dual decarboxylative cross-coupling. | ||
A comparative decarboxylative fluorination under the conditions described by MacMillan was then explored, revealing the ability of acridinium salts to complement polypyridyl transition metal systems and the organic 4-CzIPN photocatalyst (78 to 79, Scheme 21).48 Originally catalysed by a ruthenium complex in the seminal report by Yoon, the [3+2]-cycloaddition providing dihydrobenzofuran 82 further confirmed the value of the acridinium catalyst library.49
 | ||
| Scheme 21 Benchmark reactions. | ||
Another validation of the utility of the modular acridinium salt assembly was obtained by the photodeuteration of clomipramine, developed by the MacMillan group.50 Interestingly, a high selectivity for aliphatic positions was achieved with an average of 4 deuterium atoms per molecule, using a relatively low catalyst loading (1 mol%, Scheme 22).
 | ||
| Scheme 22 Deuteration of clomipramine catalysed by an acridinium salt. | ||
The range of redox properties obtained by modulating the structure of aminoacridinium catalysts hence allows to employ a common structural motif for a great variety of visible-light photoredox catalysed processes. Interestingly, a surprisingly high inherent photostability was found for the aminoacridinium salts. This property is likely required to design photocatalytic procedures while in other cases, tracing the fate of the photocatalyst during catalysis is expected to reveal interesting activation paths of utilized catalysts before a closed, productive cycle is entered. Besides the redox properties and stabilities, the kinetics, life time and energy of the excited states are other key parameters essential to refine photocatalytic reactions.
7. Conclusions
In recent years, a growing library of acridinium catalysts has been developed and utilized for various visible-light photocatalytic processes. This allowed tuning of the redox properties to a most suitable range for preparative photosynthetic applications. Similar to the polypyridyl transition-metal catalysts, amino substituents tend to lower the E1/2(P*/P−) by approximately 0.3–0.4 V while OMe groups typically allow fine tuning of the properties by 0.1–0.2 V. Moreover, alteration of substitution patterns also strongly influences the redox behaviour and thus allows further modification. Considering the numerous parameters contributing to the efficiency of the photocatalytic processes and their simultaneous variation during optimisation, it appears beneficial to employ catalysts of a common structural class to more rationally develop and improve the catalytic performance.Conflicts of interest
The authors declare no conflict of interest. Some of the described acridinium and aminoacridinium catalysts are part of a filed patent (C. Fischer, C. Sparr. WO2019057451) licenced to Solvias. The aminoacridinium catalysts will soon be commercially available.Acknowledgements
We gratefully acknowledge the Swiss National Science Foundation (BSSGI0-155902/1 and 175746), the University of Basel and the NCCR Molecular Systems Engineering for financial support. We thank Prof. M. Mayor for equipment usage, Prof. O. Wenger and Dr C. Kerzig for fruitful discussions, and Thomas Buchholz and Dragan Miladinov for skilful experimental support.Notes and references
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- K. N. Lee and M. Y. Ngai, Chem. Commun., 2017, 53, 13093–13112 RSC.
- J. Twilton, C. C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059–6071 RSC.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601–603 RSC.
- M. Brasholz, Acridinium Dyes and Quinones in Photocatalysis in Science of Synthesis, ‘Photocatalysis in Organic Synthesis’, 2018.
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed.
- T. Tsudaka, H. Kotani, K. Ohkubo, T. Nakagawa, N. V. Tkachenko, H. Lemmetyinen and S. Fukuzumi, Chem. – Eur. J., 2017, 23, 1306–1317 CrossRef CAS.
- S. Fukuzumi, K. Doi, A. Itoh, T. Suenobu, K. Ohkubo, Y. Yamada and K. D. Karlin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15572–15577 CrossRef CAS PubMed.
- M. Hoshino, H. Uekusa, A. Tomita, S. Y. Koshihara, T. Sato, S. Nozawa, S. I. Adachi, K. Ohkubo, H. Kotani and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 4569–4572 CrossRef CAS.
- A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L. C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS.
- C. Fischer and C. Sparr, Angew. Chem., Int. Ed., 2018, 57, 2436–2440 CrossRef CAS PubMed.
- A. R. White, L. Wang and D. A. Nicewicz, Synlett, 2019, 827–832 CAS.
- A. Gini, M. Uygur, T. Rigotti, J. Alemán and O. García Mancheño, Chem. – Eur. J., 2018, 24, 12509–12514 CrossRef CAS.
- A. Gini, T. Rigotti, R. Pérez-Ruiz, M. Uygur, R. Mas-Ballesté, I. Corral, L. Martínez-Fernández, V. A. de la Peña O’Shea, O. García Mancheño and J. Alemán, ChemPhotoChem, 2019, 609–612 CAS.
- F. Wu, L. Wang, J. Chen, D. A. Nicewicz and Y. Huang, Angew. Chem., Int. Ed., 2018, 57, 2174–2178 CrossRef CAS PubMed.
- L. Wang, F. Wu, J. Chen, D. A. Nicewicz and Y. Huang, Angew. Chem., Int. Ed., 2017, 56, 6896–6900 CrossRef CAS PubMed.
- J. B. McManus, N. P. R. Onuska and D. A. Nicewicz, J. Am. Chem. Soc., 2018, 140, 9056–9060 CrossRef CAS.
- K. A. Margrey, W. L. Czaplyski, D. A. Nicewicz and E. J. Alexanian, J. Am. Chem. Soc., 2018, 140, 4213–4217 CrossRef CAS.
- N. E. S. Tay and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 16100–16104 CrossRef CAS.
- J. B. McManus and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 2880–2883 CrossRef CAS PubMed.
- J. D. Griffin, C. L. Cavanaugh and D. A. Nicewicz, Angew. Chem., Int. Ed., 2017, 56, 2097–2100 CrossRef CAS PubMed.
- H. Liu, L. Ma, R. Zhou, X. Chen, W. Fang and J. Wu, ACS Catal., 2018, 8, 6224–6229 CrossRef CAS.
- K. C. Cartwright and J. A. Tunge, ACS Catal., 2018, 8, 11801–11806 CrossRef CAS.
- H. Cao, H. Jiang, H. Feng, J. M. C. Kwan, X. Liu and J. Wu, J. Am. Chem. Soc., 2018, 140, 16360–16367 CrossRef CAS PubMed.
- J. Davies, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 13361–13365 CrossRef CAS PubMed.
- L. Capaldo, R. Riccardi, D. Ravelli and M. Fagnoni, ACS Catal., 2018, 8, 304–309 CrossRef CAS.
- L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 16219–16223 CrossRef CAS.
- E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744–748 CrossRef CAS PubMed.
- J. Sim, M. W. Campbell and G. A. Molander, ACS Catal., 2019, 9, 1558–1563 CrossRef CAS.
- W. Chen, Z. Huang, N. E. S. Tay, B. Giglio, M. Wang, H. Wang, Z. Wu, D. A. Nicewicz and Z. Li, Science, 2019, 364, 1170–1174 CrossRef CAS PubMed.
- G. Zhang, X. Hu, C. W. Chiang, H. Yi, P. Pei, A. K. Singh and A. Lei, J. Am. Chem. Soc., 2016, 138, 12037–12040 CrossRef CAS PubMed.
- G. Zhang, C. Liu, H. Yi, Q. Meng, C. Bian, H. Chen, J. X. Jian, L. Z. Wu and A. Lei, J. Am. Chem. Soc., 2015, 137, 9273–9280 CrossRef CAS PubMed.
- L. Niu, J. Liu, H. Yi, S. Wang, X. A. Liang, A. K. Singh, C. W. Chiang and A. Lei, ACS Catal., 2017, 7, 7412–7416 CrossRef CAS.
- X. Hu, G. Zhang, F. Bu and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 1286–1290 CrossRef CAS PubMed.
- G. Zhang, Y. Lin, X. Luo, X. Hu, C. Chen and A. Lei, Nat. Commun., 2018, 9, 1225 CrossRef PubMed.
- S. Kato, Y. Saga, M. Kojima, H. Fuse, S. Matsunaga, A. Fukatsu, M. Kondo, S. Masaoka and M. Kanai, J. Am. Chem. Soc., 2017, 139, 2204–2207 CrossRef CAS PubMed.
- L. Huang and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 10333–10337 CrossRef CAS PubMed.
- D. Uraguchi, M. Torii and T. Ooi, ACS Catal., 2017, 7, 2765–2769 CrossRef CAS.
- C. Fischer and C. Sparr, Tetrahedron, 2018, 74, 5486–5493 CrossRef CAS.
- C. Fischer, C. Kerzig, B. Zilate, O. S. Wenger and C. Sparr, ACS Catal., 2020, 10, 210–215 CrossRef CAS.
- B. Zilate, C. Fischer, L. Schneider and C. Sparr, Synthesis, 2019, 4359–4365 CAS.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS.
- S. Ventre, F. R. Petronijevic and D. W. C. Macmillan, J. Am. Chem. Soc., 2015, 137, 5654–5657 CrossRef CAS PubMed.
- T. R. Blum, Y. Zhu, S. A. Nordeen and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 11056–11059 CrossRef CAS PubMed.
- Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. Macmillan, Science, 2017, 1187, 1182–1187 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |