
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New structural approach to rationalize the foam film stability of oppositely charged polyelectrolyte/surfactant mixtures†
Martin
Uhlig
 ab,
Oliver
Löhmann‡
c,
Salomé
Vargas Ruiz
a,
Imre
Varga
d,
Regine
von Klitzing
*ac and
Richard A.
Campbell
*ef
ab,
Oliver
Löhmann‡
c,
Salomé
Vargas Ruiz
a,
Imre
Varga
d,
Regine
von Klitzing
*ac and
Richard A.
Campbell
*ef
aStranski-Laboratorium, Technische Universität Berlin, Berlin, Germany
bFraunhofer Center for Applied Nanotechnology (CAN), Hamburg, Germany
cPhysics Department, Technische Universität Darmstadt, Darmstadt, Germany. E-mail: klitzing@smi.tu-darmstadt.de
dInstitute of Chemistry, Eötvös Loránd University, Budapest, Hungary
eInstitut Laue-Langevin, 71 avenue des Martyrs, Grenoble, France
fDivision of Pharmacy and Optometry, University of Manchester, Manchester, UK. E-mail: richard.campbell@manchester.ac.uk
First published on 20th December 2019
Abstract
A novel structural framework is presented to rationalize the foam film stability of polyelectrolyte/surfactant mixtures using neutron reflectivity data. Provision of electrostatic or steric stabilization in thin foam films is related to the spatial distributions of molecules interacting from opposing air/water interfaces. The advance is discussed in the context of many studies on mixed systems over two decades that focused on macroscopic properties such as the surface tension, elasticity, potential and composition, but for which no robust correlations have been established. This concept can now be broadened to other colloidal dispersions of high impact for technical, environmental and life science applications.
Liquid foams are nonequilibrium dispersions of gas in a liquid that are stabilized against drainage and collapse by surface active agents.1,2 Foams are of huge importance as they are used in a wide range of applications such as oil recovery, firefighting, foods and personal care products.3–5 The stability of foams is important as it determines the lifetime of the product. One way to gain insight into the stability of a foam, complementary to studies of foamability,6,7 is to study the behavior of the actual single building blocks of foams, i.e., thin foam films. The stability of thin foam films has been traditionally discussed in terms of interactions of molecules self-assembled at opposing air/water interfaces on both sides of a thin film bulk required to inhibit draining and collapse.8 This topic has been the subject of extensive experimental investigations over a prolonged period.9–13 Indeed direct relationships have been established between high foam film stability of pure surfactant solutions and low surface tension, high surface elasticity and high surface potential.8,14,15
Two stabilizing mechanisms in thin foam films are electrostatic repulsion arising from like surface charges at opposing air/water interfaces (common black films) and steric repulsion arising from factors including entanglement (Newton black films).16,17 Indeed a transition from electrostatic to steric stabilization has been described in a single system involving block co-polymer solutions with changing ionic strength.18
Other common ways to stabilize foams is through the use of proteins and nanoparticles19–21 as well as oppositely charged polyelectrolyte/surfactant (P/S) mixtures.22,23 P/S layers at the air/water interface have been extensively investigated over the last two decades, and a simple picture of the interfacial structure comprises an adsorbed layer of surfactant molecules (hydrophobic effect) with solvated polyelectrolyte bound to their head groups (electrostatic interactions).24 Even so, extended and even multilayer structures have been shown to form,25,26 although currently predictive power for their formation is lacking,27 and structural aspects have not been related systematically to foam film stability. Further, in spite of many attempts by various authors, no robust correlations between the foam film stability and the surface tension,6,10–12,28 surface elasticity,6,12,28 surface potential12,13,28 or the surface composition29 have been established for P/S mixtures.
In the present work, we examine mixtures of sulfonated poly(phenylene sulfone) (sPSO2-220) with tetradecyltrimethyl-ammonium bromide (C14TAB). It is an interesting system as the polyelectrolyte has a rather rigid conformation and high dissociation degree;30 effects of polymer rigidity have indeed been extensively investigated in the literature.31,32 The surfactant concentration is fixed in the present work at [S] = 10−4 M while the polyelectrolyte concentration is varied in the range [P] = 10−5 to 3 × 10−3 (mono)M, where (mono)M is the molar concentration of polyelectrolyte monomers. The reason for this choice is to see if different interfacial structures form with strongly varying [P] and ionic strength at fixed [S], and then establish any links to the foam film stability. It may be noted that the reason to choose such a low value of [S] was that time-dependent, nonequilibrium bulk precipitation could be discounted, even though the presence of some aggregates in the samples with low [P] could not be ruled out;33 this point is elaborated with bulk measurements of the zeta potential and optical density in part 1 of the ESI† with further experimental details found in part 2.
Characterization of macroscopic properties at the air/water interface (surface tension and elasticity) has been carried out previously in relation to disjoining pressure isotherms measured on a thin film pressure balance (TFPB).28 Here the maximum disjoining pressure is defined as the maximum pressure that can be achieved before a foam film ruptures, and it is taken as a measure of mechanical stability. From these TFPB measurements, values of the surface potential were also calculated in ref. 28 using the nonlinear Poisson–Boltzmann equation on the basis of a strong assumption about the layer structure; its validity is discussed below.
Fig. 1 summarizes the reported results for this system from the maximum disjoining pressure, surface tension and surface elasticity.28 Three different regimes of surface properties are evident. At low values of [P] (defined as regime I), foam films are only slightly stable, as shown by the low values of the maximum disjoining pressure. At intermediate values of [P] (regime II), foam films could not be formed at all and hence are least stable, yet the values of the surface tension and elasticity are very similar to those in regime I. At high values of [P] (regime III), foam films are most stable, yet the highest foam film stability ([P] = 10−3 (mono)M) does not match the lowest surface tension ([P] < 10−4 (mono)M) nor the highest surface elasticity ([P] = 2 × 10−4 (mono)M). Rationalization of foam film stability from surface tension and elasticity data alone for this system has therefore not been achieved.
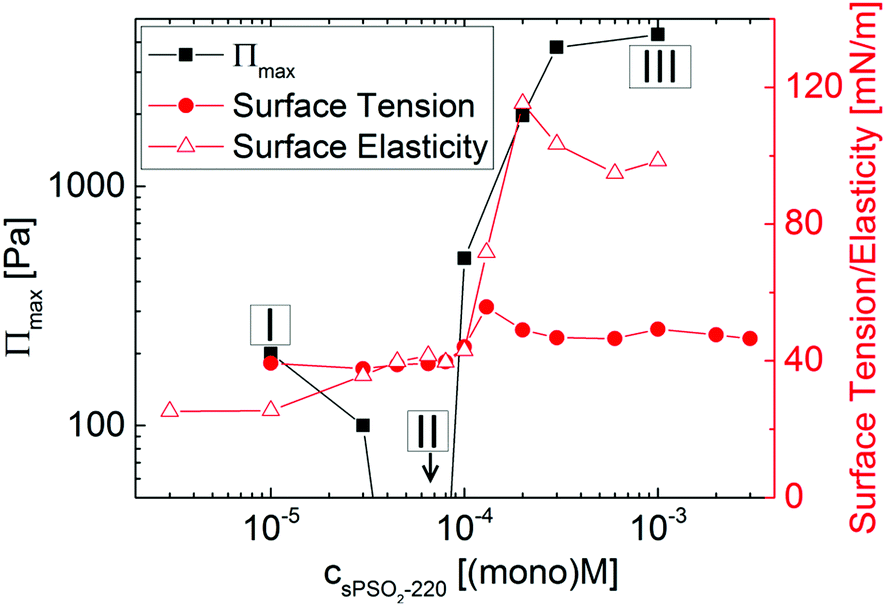 | ||
| Fig. 1 Maximum disjoining pressure, Πmax, surface tension and surface elasticity (surface dilatational modulus at 0.1 Hz) of sPSO2-220/C14TAB mixtures with varying [P] at a fixed [S] = 10−4 M; data reproduced from ref. 28. Three compositional regimes of (I) slightly stable, (II) least stable and (III) most stable foam films are marked; the data point indicated with an arrow below the displayed range corresponds to 0 Pa. | ||
The scope of the present work is to see for the first time if a correlation can be established between the foam film stability of an oppositely charged P/S mixture, varied through changes in its bulk composition, and the structures present at the air/water interface. Our aims are to resolve not only the surface composition of sPSO2-220/C14TAB mixtures, but also the locations of the components for interpretation in the context of interactions between two opposing interfaces separated by a thin film bulk. These aims are achieved through two executions of neutron reflectometry (NR) involving different isotopic contrasts and momentum transfer ranges: one accurately sensitive to the surface composition34 (i.e. the surface excesses of polyelectrolyte, ΓsPSO2-220, and surfactant, ΓC14TAB), and the other to the interfacial structure (i.e. the thickness, composition and solvation of stratified layers). Both exploit isotopic substitution where ACMW is a mixture of 8.1% D2O in H2O called air contrast matched water; more details and examples of data can be found in part 3 of the ESI.†
We start by describing results from the first execution of NR to resolve the interfacial composition. Resulting values of ΓsPSO2-220 and ΓC14TAB (left axis) and the stoichiometric ratio ΓC14TAB/ΓsPSO2-220 (right axis) are shown in Fig. 2.
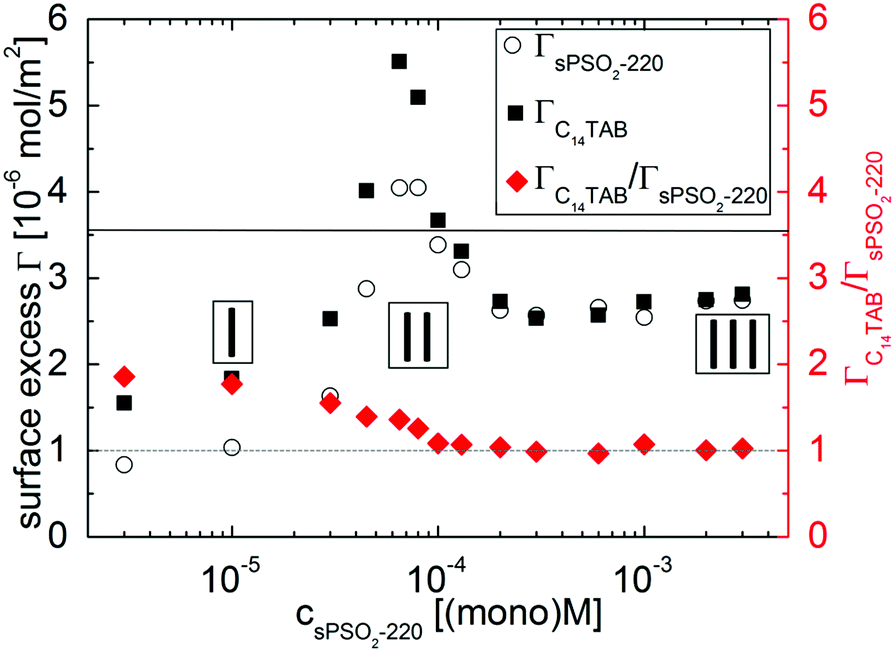 | ||
Fig. 2 Surface excesses of sPSO2-220 and C14TAB, where the full horizontal line corresponds to the surface excess for 3.5 × 10−3 M C14TAB solution (cmc),25 and the surface excess ratio ΓC14TAB/ΓsPSO2-220, where the dotted line corresponds to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio as a guide to the eye. The three compositional regimes from Fig. 1 are again marked. :1 ratio as a guide to the eye. The three compositional regimes from Fig. 1 are again marked. | ||
In regime I, there is about half a full monolayer of surfactant and less polyelectrolyte than the 1:1 interfacial charge binding expected for flexible polyelectrolytes.24 These observations can be attributed to the high rigidity of sPSO2-220, with its intrinsic persistence length of 200 Å,28 which reduces its conformational ability to bind to the surfactant head groups at the interface.
In regime II, the surface excesses of both components increase to maximum values. This effect can be linked to the higher number of bulk complexes in the system. Here ΓC14TAB is even higher than its limiting surface coverage at the critical micelle concentration (cmc) in the absence of polyelectrolyte (solid line in Fig. 2). This result implies that the interfacial structure may be more extended than the simple picture of a surfactant monolayer with polyelectrolyte bound to the head groups, i.e., that surfactant is also present beneath the surface monolayer.
In regime III, the ΓsPSO2-220 and ΓC14TAB values decrease until they become approximately constant with a 1:1 interfacial P/S ratio above [P] = 2 × 10−4 (mono)M. This trend is consistent with the reduced surface activity associated with the higher charge and greater hydrophilicity of the bulk complexes. Also, the ionic strength of the system increases markedly with changing [P] as there is no added inert electrolyte. Evidently there is no direct correlation between the foam film stability and surface composition. The changes in the data do imply, however, that different interfacial structures are formed for this system.
In order to examine the structural distribution of the components at the interface, we performed a second execution of NR that is sensitive to the location, amounts and solvation of each component for each of the three compositional regimes; details of the applied models and a list of fitting parameters can be found in part 3 of the ESI.†Fig. 3 shows NR data, model fits, scattering length density profiles and volume fraction profiles recorded for the samples as well as schematic sketches of air/water interfaces opposing each other at both sides of a thin foam film to illustrate the possible interactions.
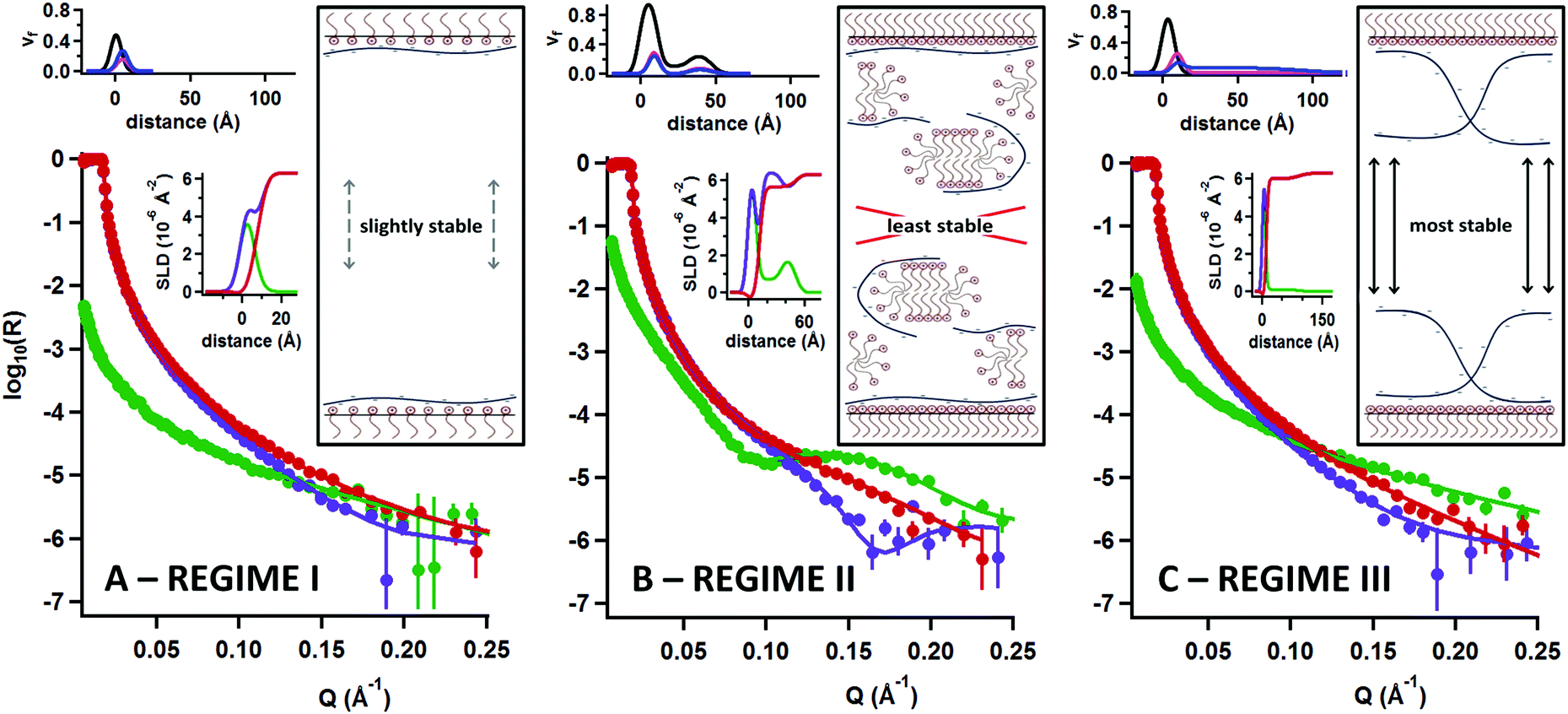 | ||
| Fig. 3 Neutron reflectivity profiles in three different isotopic contrasts of sPSO2-220 with: deuterated C14TAB in ACMW (green), deuterated C14TAB in D2O (purple) and C14TAB in D2O (red), and model fits. Data are shown in regimes I, II and III, respectively, for (A) [P] = 1 × 10−5, (B) [P] = 8 × 10−5 and (C) [P] = 3 × 10−3 (mono)M all with [S] = 10−4 M; scattering length density and volume fraction depth profiles of the model fits are shown as insets where in the latter case the species are: (black) surfactant chains, (pink) surfactant head groups and (blue) polyelectrolyte; sketches of possible structures at opposing air/water interfaces of a thin foam film are also shown (counterions are omitted for clarity due to insensitivity of the technique to their presence). | ||
First, in regime I (Fig. 3A), the optimized structural model consists of a layer of C14TAB molecules with their chains facing the air, and a thin layer of sPSO2-220 bound to the solvated head groups. The total thickness is <10 Å. Although there is a small excess of surfactant (i.e. ΓC14TAB > ΓsPSO2-220), excess charge at the interface would provide only weak double layer repulsion to the thin foam film. As such, drainage and collapse would be only poorly inhibited by electrostatic stabilization. It follows that these foam films are only slightly stable.
Next in regime II (Fig. 3B), where greater than monolayer coverage of surfactant was observed above, there is a pronounced Kiessig fringe in the data of the two contrasts involving deuterated surfactant. The fringe position demonstrates unambiguously that surfactant is present in an extended structure with an average depth of ∼50 Å (Bragg's law). The optimized structural model consists of a C14TAB monolayer with a compact layer of sPSO2-220 bound to the surfactant head groups, then a low coverage of a C14TAB bilayer (or hemimicelles), and lastly an additional outer mixed layer. This general structure is clearly resolved from the data, and was reproduced in different experiments. Even so, some details of the structure remain unclear. The coverage of the extended structure varies from 10–40% between the isotopic contrasts measured, which we attribute to isotope-specific effects related to the difference in hydrogen bonding of CH and CD bonds. Also, we have sketched the inner layer intuitively as a matrix of polyelectrolyte associated with hemimicelles, but further work would be required to refine this physical picture. As the bulk surfactant concentration is a factor of 30 less than the cmc of the pure surfactant, the extended structure must be either templated by the interface or delivered by any aggregates present in the bulk that interact with the surface. As such, the mixed inner layers that face each other in a thin foam film would not inhibit drainage effectively due to a lack of electrostatic stabilization from opposing charged planes or steric stabilization from entangled polyelectrolyte chains alone. It follows that these foam films are least stable and result in rapid collapse.
Lastly, in regime III (Fig. 3C), although the data look quite similar to those of regime I, the falloff of the data of the contrast involving hydrogenous surfactant in D2O is greater, implying that the polyelectrolyte layer is thicker. The optimized structural model, like in regime I, again consists of a C14TAB layer with sPSO2-220 bound to the surfactant head groups, but in this case indeed the polyelectrolyte adopts an extended conformation with a layer thickness of 80 Å and a volume fraction of <10%. There is 1:1 stoichiometry between the bound polyelectrolyte and surfactant monolayer, indicating that double layer repulsion between opposing interfaces is not the origin of the high foam film stability. At the same time, the thickness of these foam films has been determined as ∼350 Å,28 which means that in addition to the polyelectrolyte bound to surfactant at the interface, there is a contribution of polyelectrolyte present in the thin film bulk estimated as a further ∼3%. In a thin foam film, the extended polyelectrolyte layer at the interface and free coils in the bulk can interpenetrate giving rise to high local segment densities, which in turn results in high osmotic pressure. As such, the repulsive forces necessary to inhibit drainage would be provided primarily by steric stabilization as a result of the dynamic entanglement of extended polyelectrolyte chains. It follows that these foam films are the most stable.
An outstanding curiosity is that high calculated values of the surface potential in the original work on this system coincide with the most stable foam films of regime III.28 These calculations were derived from fits to disjoining pressure isotherms using the nonlinear Poisson–Boltzmann equation, which is based on the model assumption that charges at opposing interfaces of a thin foam film are located in a defined plane. Indeed a correlation between foam film stability and surface potential has been established for pure surfactant solutions.8,14,15 However, in the present work two important points arise for the samples in regime III: first, the interface consists of 1:1 binding of the two components (Fig. 2), so it is not logical that the surface potential is highest; second, the polyelectrolyte layer extends into the thin film bulk beneath the surfactant monolayer (Fig. 3C) so the assumption that charges are present in a defined plane is not valid. It is clear that application of the described method to calculate the surface potential for mixed systems can in certain cases therefore be physically unjustified, which reinforces the value of structural data like those resolved using NR in the present work.
The novel framework presented to rationalize the foam film stability of oppositely charged P/S mixtures focuses, for the first time, on the structural distribution of components that face the thin film bulk. Different conformations at two opposing air/water interfaces of a thin foam film are rationalized in terms of established stabilization mechanisms – electrostatic and steric repulsion – needed to inhibit drainage. The significance of this advance must be placed in the context of the large number of studies that aimed to relate foam film stability of mixed P/S systems to the surface tension,6,10–12,28 elasticity,6,12,28 potential12,13,28 and composition,23 but for which exceptions to the behavior of pure surfactant solutions have been repeatedly observed.
This study lays the foundations for work on a range of systems, such as those involving proteins and nanoparticles. Tests on different systems of the new framework presented are now required to assess its generality, and to explore its application in the development of new materials such as smart foams,35 which have great promise to act as more efficient foam products in the future.
We thank the ILL for time on FIGARO (DOI: 10.5291/ILL-DATA.9-12-381), Simon Wood for technical support, and the group of Klaus-Dieter Kreuer (Max Planck Institute for Solid State Research, Stuttgart) for the sPSO2-220. MU thanks the DFG for financial support from SPP “Kolloid- und Verfahrenstechnik” 1273 Kl-1165/10. The work was also supported by the Hungarian National Research, Development and Innovation Office (K116629). The open access fee was covered by FILL2030, a European Union project within the European Commission's Horizon 2020 Research and Innovation programme under grant agreement N°731096.
Conflicts of interest
There are no conflicts to declare.References
- G. Gochev, Curr. Opin. Colloid Interface Sci., 2015, 20, 115 CrossRef CAS.
- J. Bae, K. Lee, S. Seo, J. G. Park, Q. Zhou and T. Kim, Nat. Commun., 2019, 10, 3209 CrossRef PubMed.
- J. M. Rodríguez Patino, C. Carrera Sánchez and M. R. Rodríguez Niño, Adv. Colloid Interface Sci., 2008, 140, 95 CrossRef PubMed.
- E. Rio, W. Drenckhan, A. Salonen and D. Langevin, Adv. Colloid Interface Sci., 2014, 205, 74 CrossRef CAS PubMed.
- C.-Y. Lai, B. Rallabandi, A. Perazzo, Z. Zheng, S. E. Smiddy and H. A. Stone, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8082 CrossRef CAS PubMed.
- R. Petkova, S. Tcholakova and N. D. Denkov, Colloids Surf., A, 2013, 438, 174 CrossRef CAS.
- J. Wang, A. V. Nguyen and S. A. S. Farrokhpay., Adv. Colloid Interface Sci., 2016, 228, 55 CrossRef CAS PubMed.
- D. Exerowa, N. V. Churaev, T. Kolarov, N. E. Esipova, N. Panchev and Z. M. Zorin, Adv. Colloid Interface Sci., 2003, 104, 1 CrossRef CAS PubMed.
- V. Bergeron, Langmuir, 1997, 13, 3474 CrossRef CAS.
- B. M. Folmer and B. Kronberg, Langmuir, 2000, 16, 5987 CrossRef CAS.
- C. Monteux, C. E. Williams, J. Meunier, O. Anthony and V. Bergeron, Langmuir, 2004, 20, 57 CrossRef CAS PubMed.
- N. Kristen, V. Simulescu, A. Vüllings, A. Laschewsky, R. Miller and R. v. Klitzing, J. Phys. Chem. B, 2009, 113, 7986 CrossRef CAS PubMed.
- H. Fauser and R. v. Klitzing, Soft Matter, 2014, 10, 6903 RSC.
- C. Stubenrauch and R. v. Klitzing, J. Phys.: Condens. Matter, 2003, 1198, R1197 CrossRef.
- C. Stubenrauch and R. Miller, J. Phys. Chem. B, 2004, 108, 6412 CrossRef CAS PubMed.
- D. Exerowa, A. Nikolov and M. Zacharieva, J. Colloid Interface Sci., 1981, 81, 419 CrossRef.
- J. Liu, G. Xu, S. Yuan, P. Jiang. and J. Dispers, Sci. Tech., 2003, 24, 779 CAS.
- D. Exerowa, R. Sedev, R. Ivanova, T. Kolarov and T. F. Tadros, Colloids Surf., A, 1997, 123–124, 277 CrossRef CAS.
- M. Chen, G. Sala, M. B. J. Meinders, H. J. F. van Valenberg, E. van der Linden and L. M. C. Sagis, Colloids Surf., B, 2017, 149, 56 CrossRef CAS PubMed.
- T. M. Tenhunen, T. Pohler, A. Kokko, H. Orelma, M. Schenker, P. Gane and T. Tammelin, Nanomaterials, 2018, 8, 651 CrossRef PubMed.
- N. Hu, Z. L. Wu, L. X. Jin, Z. W. Li, W. Liu, D. Huang and C. Y. Yang, Sep. Purif. Technol., 2019, 209, 392 CrossRef CAS.
- A. Bureiko, A. Trybala, N. Kovalchuk and V. Starov, Adv. Colloid Interface Sci., 2014, 222, 670 CrossRef PubMed.
- F. Schulze-Zachau and B. Braunschweig, Langmuir, 2017, 33, 3499 CrossRef CAS PubMed.
- A. Asnacios, D. Langevin and J.-F. Argillier, Macromolecules, 1996, 29, 7412 CrossRef CAS.
- D. J. F. Taylor, R. K. Thomas and J. Penfold, Langmuir, 2002, 18, 4748 CrossRef CAS.
- R. A. Campbell, M. Yanez Arteta, A. Angus-Smyth, T. Nylander and I. Varga, J. Phys. Chem. B, 2012, 116, 7981 CrossRef CAS PubMed.
- P. Li, J. Penfold, R. K. Thomas and H. Xu, Adv. Colloid Interface Sci., 2019, 269, 43 CrossRef CAS PubMed.
- M. Uhlig, R. Miller and R. v. Klitzing, Phys. Chem. Chem. Phys., 2016, 18, 18414 RSC.
- H. Fauser, R. v. Klitzing and R. A. Campbell, J. Phys. Chem. B, 2015, 119, 348 CrossRef CAS PubMed.
- M. Schuster, C. C. de Araujo, V. Atanasov, H. T. Andersen, K.-D. Kreuer and J. Maier, Macromolecules, 2009, 42, 3129 CrossRef CAS.
- H. Ritacco, P.-A. Albouy, A. Bhayyacharyya and D. Langevin, Phys. Chem. Chem. Phys., 2000, 2, 5243 RSC.
- F. Kleinschmidt, C. Stubenrauch, J. Delacotte, R. v. Klitzing and D. Langevin, J. Phys. Chem. B, 2009, 113, 3972 CrossRef CAS PubMed.
- I. Varga and R. A. Campbell, Langmuir, 2017, 33, 5915 CrossRef CAS PubMed.
- R. A. Campbell, A. Tummino, B. A. Noskov and I. Varga, Soft Matter, 2016, 12, 5304 RSC.
- M. Schnurbus, L. Stricker, B. J. Ravoo and B. Braunschweig, Langmuir, 2018, 34, 6028 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Zeta potential and UV-visible spectroscopy data, further experimental details and NR data evaluation. See DOI: 10.1039/c9cc08470c |
| ‡ Current address: European Spallation Source ESS ERIC, P. O. Box 176, Lund, Sweden. |
| This journal is © The Royal Society of Chemistry 2020 |