
Geometric effect of Au nanoclusters on room temperature CO oxidation†

Abstract
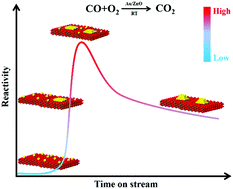
We report the effect of in situ transforming ZnO supported single Au atoms to 3-dimensional Au clusters (Au3D) on room temperature CO oxidation activity. We discovered that an intermediate, highly distorted 2-dimensional Au layer species is much more active than both the Au3D clusters and the Au single atoms. The geometric arrangement of Au clusters and their interactions with support surfaces play a pivotal role in determining their catalytic performance in room temperature CO oxidation on ZnO supported Au catalysts.
 Please wait while we load your content...
Please wait while we load your content...

