
Photoinduced deaminative strategies: Katritzky salts as alkyl radical precursors

Abstract
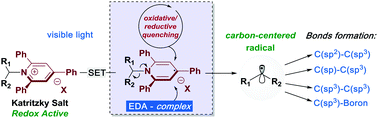
Primary amines are one of the most predominant functional groups found in organic molecules. These entities help form the chemical architecture of natural products, bioactive molecules, synthetic building blocks and catalysts. Due to their ubiquitous presence, the development of strategies for the construction of C–C or C–X bonds through deaminative processes is of high importance. Deaminative methods offer new possibilities on the retrosynthetic rationale, and enable late-stage-functionalization of complex structures. As a result of the recent development of photoinduced processes, a variety of photo-mediated deaminative protocols employing 2,4,6-triphenyl-pyridinium salts – Katritzky Salts – as activating agents have been recently realized. This review covers the most recent developments of deaminative strategies by using Katritzky Salts as alkyl radical reservoirs, with particular concern on photoinduced processes applied to organic synthesis.
- This article is part of the themed collections: Celebrating Latin American Talent in Chemistry, Organic Synthesis and Most popular organic chemistry articles
 Please wait while we load your content...
Please wait while we load your content...

