
Protein degradation through covalent inhibitor-based PROTACs†
Gang
Xue
,
Jiahui
Chen
,
Lihong
Liu
,
Danli
Zhou
,
Yingying
Zuo
,
Tiancheng
Fu
and
Zhengying
Pan
 *
*
State Key Laboratory of Chemical Oncogenomics, Key Laboratory of Chemical Genomics, Engineering Laboratory for Chiral Drug Synthesis, School of Chemical Biology and Biotechnology Shenzhen Graduate School, Peking University, Xili University Town, PKU Campus, F-311, Shenzhen, 518055, China. E-mail: panzy@pkusz.edu.cn
First published on 10th January 2020
Abstract
Tremendous advancements in proteolysis targeting chimera (PROTAC) technology have been made in recent years. However, whether a covalent inhibitor-based PROTAC can be developed remains controversial. Here, we successfully developed chimeric degraders based on covalent inhibitors to degrade BTK and BLK kinases, demonstrating that covalent inhibitor-based PROTACs are viable and useful tools.
Targeted protein degradation has been used in the clinic to treat patients by using monovalent small molecule degraders, such as fulvestrant, lenalidomide and pomalidomide.1 Proteolysis targeting chimera (PROTAC) technology emerged by using bivalent chemical agents in 2001,2 and a PROTAC molecule is composed of a protein binding ligand, an E3 ubiquitin ligase ligand and a linker connecting the two ligands. PROTACs induce an E3 ligase to recruit the target protein, resulting in target protein polyubiquitination and degradation via the ubiquitin–proteasome system. Degradation of the target protein by PROTACs is normally considered as an event-driven pharmacological effect and acts in a catalytic manner.3 In recent years, PROTACs have made great progress and have been successfully applied to degrade various kinds of proteins,4 including kinases, nuclear receptors, BET proteins, and other proteins.
Covalent inhibitors have experienced a resurgence during the past decade both as chemical tools and as approved medicines. We have been interested in combining these two powerful modalities to generate covalent inhibitor-based PROTACs. In the literature, the degradation results of covalent PROTACs are mixed. The first PROTAC article described a covalent inhibitor-based degrader but was only tested in an in vitro system.2 Additional reports on covalently binding PROTACs have indicated that such molecules degrade proteins in cells.5–7 However, a recent report clearly showed that a covalent PROTAC could not induce the degradation of its target protein – Bruton's tyrosine kinase (BTK).8 Here we present our efforts to develop covalent inhibitor-based PROTACs that successfully degrade BTK and B lymphocyte kinase (BLK) in live cells.
BTK is a non-receptor cytoplasmic tyrosine kinase9 and participates in the B-cell receptor (BCR) signaling pathway, which plays a key role in B cell lymphomas.10 The first BTK covalent inhibitor, ibrutinib11 has been approved by the FDA for the treatment of various B cell malignancies. BTK PROTACs12–15 require a BTK binding moiety connected via a linker to an E3 ligase ligand. For the BTK binding moiety, the covalent inhibitors ibrutinib and PLS-12316 were selected (Chart 1 and Fig. S1, ESI†), because of their high affinities and the large structural differences between their scaffolds. Based on our previous work on converting ibrutinib17 and PLS-12318 into fluorescent BTK probes, we selected the carboxyl groups on compound 1 and compound 2 as the egress point to ligate the linkers (Chart 1). For E3 ligase ligands, pomalidomide and VH032, which recruit E3 ubiquitin ligase cereblon (CRBN) and VHL, respectively, were employed (Fig. S1, ESI†). Based on the X-ray crystal structures of CRBN complexed with pomalidomide19 (PDB ID: 4CI3) and VHL complexed with VH03220 (PDB ID: 4W9H), the linker bound to pomalidomide was ligated at carbon-4 on the phthalimide ring and the linker bound to VHL was ligated through the terminal acetamide group of VH032. Thus compounds 3–10 were designed and synthesized (Schemes S1–S4, ESI†).
 | ||
| Chart 1 Chemical structures of BTK inhibitors used in this study. | ||
We first connected the ibrutinib analogue 1 to pomalidomide with different linkers21 (3a–d, 4a–d) (Fig. 1a) and examined their ability to degrade BTK in cells. After treating K562 cells with increasing concentrations of compounds for 24 h, we analyzed cell lysates by Western blot. Decreases in BTK protein levels were observed with the compounds’ concentrations ranging from 100 nM to 3 μM, whereas ibrutinib itself did not reduce the BTK protein level (Fig. S2, ESI†). However, the level of BTK protein could only be reduced by approximately 50%. The degradation effects of the compounds (3a–d, 4a–d) are shown in Fig. 1b and c and Fig. S2 (ESI†).
 | ||
Fig. 1 The degradation of BTK by PROTACs 3–6. (a) Chemical structures of PROTACs 3 and 4. (b) Degradation of BTK by PROTACs with different linker lengths and linker compositions in K562 cells for 24 h at the two indicated concentrations. (c) BTK protein levels in cells. Numbers were calculated by the BTK/GAPDH ratio with normalization by the DMSO control as 100. (d) Chemical structures of PROTACs 5 and 6. (e) Degradation of BTK by PROTACs 5 and 6 in K562 cells for 24 h at the two indicated concentrations. (f) BTK protein levels in cells. Numbers were calculated by the BTK/GAPDH ratio with normalization by the DMSO control as 100. The bars in the graphs show the means![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ± standard deviations from two biological replicates. ± standard deviations from two biological replicates. | ||
Since the ligand-induced protein–protein interaction between the E3 ligase and target protein is very important for the degradation efficiency of PROTACs,22 we conjugated pomalidomide with another covalent inhibitor, PLS-123, whose scaffold and its binding mode with BTK were different from those of ibrutinib.23 Four analogues with different linkers (Fig. 1d) were synthesized, and their degradation activities were evaluated. PROTACs with PEG linkers (5a, 5b) did not show appreciable degradation activity (Fig. 1e). However, PROTACs 6a and 6b with different lengths of carbon chains effectively degraded BTK. PROTAC 6b had an improved degradation activity with its DC50 (the concentration causing 50% reduction in the protein level relative to the vehicle-treated sample) value likely less than 300 nM and Dmax (the maximum reduction of the protein level relative to the vehicle-treated sample)24 value of 75% at 1 μM (Fig. 1f).
Since the degradation ability of PROTACs was also impacted by the E3 ligase,25 we further replaced the pomalidomide in PROTACs 3a and 6b with VH032 to employ another E3 ligase (VHL) and synthesized PROTACs 7 and 8 (Fig. 2a). Indeed, compound 7 showed the most significant ability to reduce the BTK protein level with DC50 = 136 nM and Dmax = 88% (Fig. 2b and c). By contrast, the degradation activity of PROTAC 8 was significantly lower than that of PROTAC 6b.
 | ||
| Fig. 2 The comparison of BTK degradation by PROTACs with different E3 ligands. (a) Chemical structures of PROTACs 7 and 8. (b) Western blot results of BTK and GAPDH. K562 cells were treated with PROTACs for 18 h at the two indicated concentrations. (c) BTK protein levels in cells. Numbers were calculated by the BTK/GAPDH ratio with normalization by the DMSO control as 100. The bars in the graphs show the means ± standard deviations from three biological replicates. *P < 0.05, **P < 0.01 and ***P < 0.001 are from unpaired t-tests. | ||
Non-covalent inhibitor-based PROTACs 9 and 10 were obtained by replacing the acrylamide reactive group in PROTACs 6b and 7 with a propanamide group. Competition assays with PROTACs 6b, 7, 9 and 10 and covalent fluorescent probe 1117 (Fig. S1, ESI†) were also performed. As expected, preincubation of PROTACs 6b and 7 with BTK for 1.5 h greatly decreased the binding of probe 11 with BTK, while 9 and 10 did not (Fig. S3, ESI†). In a cellular activity assay, the treatment with PROTAC 7 inhibited BTK's autophosphorylation of Tyr223 even after 7 was washed-out with fresh medium (Fig. S4, ESI†). Additionally, compound 7 displayed an incubation time-dependent inhibition behaviour in a BTK kinase activity assay (Fig. S5, ESI†). Collectively, these results indicate that PROTACs 6b and 7 are irreversibly bound to BTK. Head-to-head comparisons were then performed to compare the degradation activities of covalent PROTACs (6b and 7) and non-covalent PROTACs (9 and 10) (Fig. 3a). The results showed that the BTK protein levels in cells treated with PROTAC 7 were significantly lower than those treated with PROTAC 9, while PROTACs 10 and 6b showed comparable results (Fig. 3b and c). The IC50 values of these compounds against BTK were also measured (Fig. 3d). Compared to PROTAC 9, PROTAC 7 is a more potent inhibitor and a better degrader, and PROTACs 10 and 6b have comparable potencies in enzymatic assays and degradation abilities in live cells. The catalytic property is an important and interesting feature of degraders; some PROTACs with weak binding moieties can still have potent degradation abilities. PROTACs with a covalent irreversible binder would likely lose this property. However, the strong binding potencies of covalent PROTACs 7 and 6b appear to compensate well for the loss of turnovers.
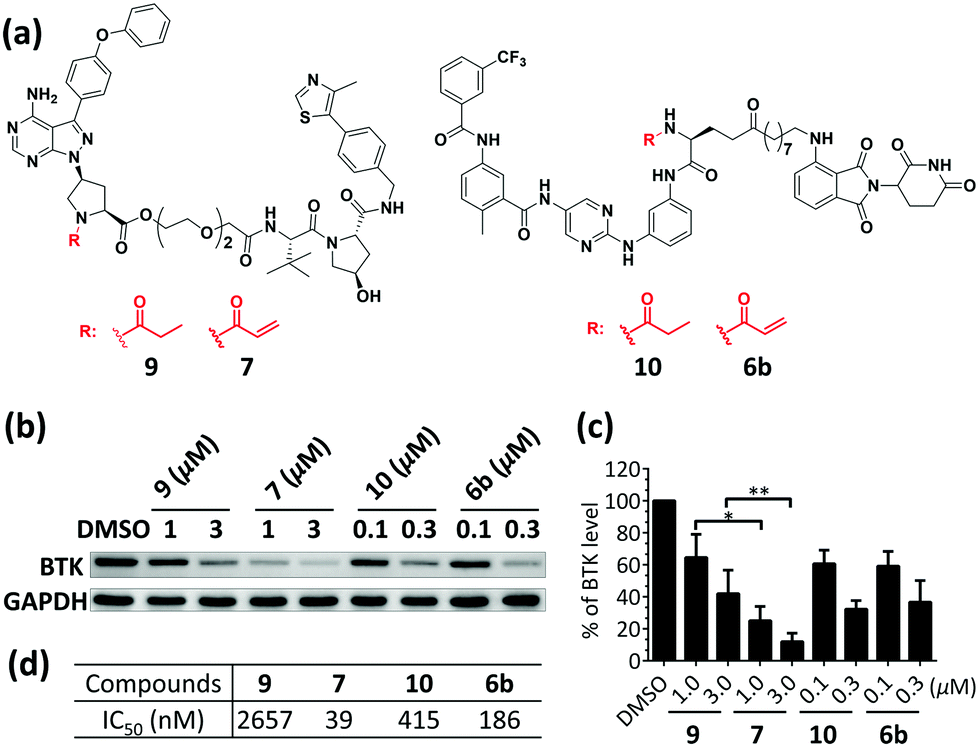 | ||
| Fig. 3 The comparison of BTK degradation by covalent and non-covalent PROTACs. (a) Chemical structures of PROTACs 6b, 7, 9, and 10. (b) Western blot results of BTK and GAPDH. K562 cells were treated with PROTACs for 18 h at the two indicated concentrations. (c) BTK protein levels in cells. Numbers were calculated by the BTK/GAPDH ratio with normalization by the DMSO control as 100. The bars in the graphs show the means ± standard deviations from three biological replicates. *P < 0.05 and **P < 0.01 are from unpaired t-tests. (d) IC50 values of PROTACs 6b, 7, 9, and 10 against BTK kinase. | ||
Since probe 1218 (Fig. S1, ESI†), which was derived from compound 2, was found to covalently bind to another kinase, BLK (Fig. S6, ESI†), we performed a competitive assay between PROTAC 7 and probe 12. Preincubation of PROTAC 7 for 2 h greatly decreased the labeling of the BLK protein, indicating that PROTAC 7 also covalently binds to BLK (Fig. 4a). Thus, we treated Ramos cells with various concentrations of PROTAC 7 for 18 h to evaluate its ability to degrade the BLK protein. Indeed, PROTAC 7 potently reduced the BLK protein level with a DC50 of 220 nM and a Dmax of 75% (Fig. 4b and Fig. S7, ESI†).
 | ||
| Fig. 4 PROTAC 7 covalently bind to and degrade BLK. (a) Competition assay between PROTAC 7 and covalent fluorescent probe 12. (b) Western blot results of BLK and GAPDH upon increasing the concentration of 7 in Ramos cells. | ||
To investigate the mechanism of BTK and BLK degradation using PROTAC 7, cells were preincubated with the E3 ligase-VHL inhibitor (VH032), the BTK and BLK inhibitor (ibrutinib), the proteasome inhibitor (carfilzomib) or the ubiquitin-activating enzyme inhibitor (MLN7243), then it was found that PROTAC 7 could not induce the degradation of BTK (Fig. S8a, ESI†) and BLK (Fig. S8b, ESI†), which indicates that the degradation requires the proteasome system and PROTAC 7's binding to the target protein and the E3 ligase-VHL.
In this study, we successfully developed PROTACs from covalent kinase inhibitors. These covalent inhibitor-based PROTACs irreversibly bound with target kinases and achieved excellent degradation potency in live cells. PROTAC 7 effectively degraded the BTK protein with a DC50 of 136 nM and a Dmax of 88% and also degraded the BLK protein with a DC50 of 220 nM and a Dmax of 75%. Thus, covalently binding to kinases does not prevent the formation of effective PROTACs. However, our optimization process also clearly indicates that all three components (ligand, linker and E3 ligase) of PROTAC molecules need to be adjusted to obtain good degraders. As covalent inhibitors with superb binding affinities toward traditionally druggable and undruggable targets have been developed and have achieved success in the clinic,10,26 our results would strongly suggest adapting them into PROTACs to further extend the scope of PROTACs.
We acknowledge funding support from the National Natural Science Foundation of China (81872749), and the Shenzhen Science and Technology Innovation Commission (JCYJ20160226105227446 and 1210318253).
Conflicts of interest
There are no conflicts to declare.References
- G. F. Watt, P. Scott-Stevens and G. Lu, Drug Discovery Today: Technol., 2019, 31, 69–80 CrossRef PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- D. P. Bondeson, A. Mares, I. E. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, W. den Besten, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Nat. Chem. Biol., 2015, 11, 611–617 CrossRef CAS PubMed.
- M. Pettersson and C. M. Crews, Drug Discovery Today: Technol., 2019, 31, 15–27 CrossRef PubMed.
- H. Lebraud, D. J. Wright, C. N. Johnson and T. D. Heightman, ACS Cent. Sci., 2016, 2, 927–934 CrossRef CAS PubMed.
- G. M. Burslem, B. E. Smith, A. C. Lai, S. Jaime-Figueroa, D. C. McQuaid, D. P. Bondeson, M. Toure, H. Dong, Y. Qian, J. Wang, A. P. Crew, J. Hines and C. M. Crews, Cell Chem. Biol., 2018, 25, 67–77.e63 CrossRef CAS PubMed.
- L. Peng, Z. Zhang, C. Lei, S. Li, Z. Zhang, X. Ren, Y. Chang, Y. Zhang, Y. Xu and K. Ding, ACS Med. Chem. Lett., 2019, 10, 767–772 CrossRef CAS PubMed.
- C. P. Tinworth, H. Lithgow, L. Dittus, Z. I. Bassi, S. E. Hughes, M. Muelbaier, H. Dai, I. E. D. Smith, W. J. Kerr, G. A. Burley, M. Bantscheff and J. D. Harling, ACS Chem. Biol., 2019, 14, 342–347 CrossRef CAS PubMed.
- C. M. Lewis, C. Broussard, M. J. Czar and P. L. Schwartzberg, Curr. Opin. Immunol., 2001, 13, 317–325 CrossRef CAS PubMed.
- R. W. Hendriks, S. Yuvaraj and L. P. Kil, Nat. Rev. Cancer, 2014, 14, 219–232 CrossRef CAS PubMed.
- Z. Pan, H. Scheerens, S. J. Li, B. E. Schultz, P. A. Sprengeler, L. C. Burrill, R. V. Mendonca, M. D. Sweeney, K. C. Scott, P. G. Grothaus, D. A. Jeffery, J. M. Spoerke, L. A. Honigberg, P. R. Young, S. A. Dalrymple and J. T. Palmer, ChemMedChem, 2007, 2, 58–61 CrossRef CAS PubMed.
- A. D. Buhimschi, H. A. Armstrong, M. Toure, S. Jaime-Figueroa, T. L. Chen, A. M. Lehman, J. A. Woyach, A. J. Johnson, J. C. Byrd and C. M. Crews, Biochemistry, 2018, 57, 3564–3575 CrossRef CAS PubMed.
- H. T. Huang, D. Dobrovolsky, J. Paulk, G. Yang, E. L. Weisberg, Z. M. Doctor, D. L. Buckley, J. H. Cho, E. Ko, J. Jang, K. Shi, H. G. Choi, J. D. Griffin, Y. Li, S. P. Treon, E. S. Fischer, J. E. Bradner, L. Tan and N. S. Gray, Cell Chem. Biol., 2018, 25, 88–99.e86 CrossRef CAS PubMed.
- Y. Sun, X. Zhao, N. Ding, H. Gao, Y. Wu, Y. Yang, M. Zhao, J. Hwang, Y. Song, W. Liu and Y. Rao, Cell Res., 2018, 28, 779–781 CrossRef CAS PubMed.
- A. Zorba, C. Nguyen, Y. Xu, J. Starr, K. Borzilleri, J. Smith, H. Zhu, K. A. Farley, W. Ding, J. Schiemer, X. Feng, J. S. Chang, D. P. Uccello, J. A. Young, C. N. Garcia-Irrizary, L. Czabaniuk, B. Schuff, R. Oliver, J. Montgomery, M. M. Hayward, J. Coe, J. Chen, M. Niosi, S. Luthra, J. C. Shah, A. El-Kattan, X. Qiu, G. M. West, M. C. Noe, V. Shanmugasundaram, A. M. Gilbert, M. F. Brown and M. F. Calabrese, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E7285–E7292 CrossRef PubMed.
- N. Ding, X. Li, Y. Shi, L. Ping, L. Wu, K. Fu, L. Feng, X. Zheng, Y. Song and Z. Pan, Oncotarget, 2015, 6, 15122–15136 Search PubMed.
- J. Chen, X. Wang, F. He and Z. Pan, Bioconjugate Chem., 2018, 29, 1640–1645 CrossRef CAS PubMed.
- Y. Zuo, Y. Shi, X. Li, Y. Teng and Z. Pan, Sci. Rep., 2015, 5, 16136 CrossRef CAS PubMed.
- E. S. Fischer, K. Bohm, J. R. Lydeard, H. Yang, M. B. Stadler, S. Cavadini, J. Nagel, F. Serluca, V. Acker, G. M. Lingaraju, R. B. Tichkule, M. Schebesta, W. C. Forrester, M. Schirle, U. Hassiepen, J. Ottl, M. Hild, R. E. Beckwith, J. W. Harper, J. L. Jenkins and N. H. Thoma, Nature, 2014, 512, 49–53 CrossRef CAS PubMed.
- C. Galdeano, M. S. Gadd, P. Soares, S. Scaffidi, I. Van Molle, I. Birced, S. Hewitt, D. M. Dias and A. Ciulli, J. Med. Chem., 2014, 57, 8657–8663 CrossRef CAS PubMed.
- B. Zhou, J. Hu, F. Xu, Z. Chen, L. Bai, E. Fernandez-Salas, M. Lin, L. Liu, C. Y. Yang, Y. Zhao, D. McEachern, S. Przybranowski, B. Wen, D. Sun and S. Wang, J. Med. Chem., 2018, 61, 462–481 CrossRef CAS PubMed.
- M. S. Gadd, A. Testa, X. Lucas, K. H. Chan, W. Chen, D. J. Lamont, M. Zengerle and A. Ciulli, Nat. Chem. Biol., 2017, 13, 514–521 CrossRef CAS PubMed.
- X. Li, Y. Zuo, G. Tang, Y. Wang, Y. Zhou, X. Wang, T. Guo, M. Xia, N. Ding and Z. Pan, J. Med. Chem., 2014, 57, 5112–5128 CrossRef CAS PubMed.
- K. H. Chan, M. Zengerle, A. Testa and A. Ciulli, J. Med. Chem., 2018, 61, 504–513 CrossRef CAS PubMed.
- A. C. Lai, M. Toure, D. Hellerschmied, J. Salami, S. Jaime-Figueroa, E. Ko, J. Hines and C. M. Crews, Angew. Chem., 2016, 55, 807–810 CrossRef CAS PubMed.
- F. McCormick, Biochem. J., 2019, 476, 365–374 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc08238g |
| This journal is © The Royal Society of Chemistry 2020 |