
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMass spectrometry reveals the assembly pathway of encapsulated ferritins and highlights a dynamic ferroxidase interface†
Jennifer
Ross
 a,
Thomas
Lambert
a,
Cecilia
Piergentili
b,
Didi
He
c,
Kevin J.
Waldron
d,
C. Logan
Mackay
a,
Jon
Marles-Wright
*b and
David J.
Clarke
*a
a,
Thomas
Lambert
a,
Cecilia
Piergentili
b,
Didi
He
c,
Kevin J.
Waldron
d,
C. Logan
Mackay
a,
Jon
Marles-Wright
*b and
David J.
Clarke
*a
aEaStCHEM School of Chemistry, The University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, Scotland, UK. E-mail: Dave.clarke@ed.ac.uk
bSchool of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: Jon.marles-wright1@ncl.ac.uk
cStructural Genomics Consortium, University of Oxford, Old Road Campus Research Building, Roosvelt Drive, Oxford, OX3 7DQ, UK
dInstitute for Cell and Molecular Biosciences, Newcastle University, Newcastle upon Tyne NE2 4HH, UK
First published on 13th February 2020
Abstract
Encapsulated ferritins (EncFtn) are a recently characterised member of the ferritin superfamily. EncFtn proteins are sequestered within encapsulin nanocompartments and form a unique biological iron storage system. Here, we use native mass spectrometry and hydrogen–deuterium exchange mass spectrometry to elucidate the metal-mediated assembly pathway of EncFtn.
Ferritins are ubiquitous enzymes used to oxidise and store iron in almost all life forms.1 In classical ferritins, a 2-iron ferroxidase centre catalyses the oxidation of Fe(II) and the resulting Fe(III) is stored within their spherical quaternary nanocage structure.2 A recently characterised sub-family of ferritins, encapsulated ferritins (EncFtn), are sequestered within 20–50 nm icosahedral encapsulin nanocompartments (Enc).3–5 EncFtn proteins are directed into the interior of the encapsulin shell by a C-terminal localisation sequence, forming an encapsulin system.3,6 These proteins work together to form a competent iron-store, with the EncFtn responsible for iron oxidation, while iron is mineralised and stored within the encapsulin nanocompartment.7,8 Due to their greater size, Enc–EncFtn systems have a much greater iron storage potential than classical ferritins and DNA-binding Protein from Starved cells (DPS) nanocages.4
Using X-ray crystallography, we have recently shown that members of the EncFtn family adopt a conserved annular quaternary structure (Fig. 1A).3,9 This is formed from a pentamer of dimers, the topology of which results in two distinct dimer interfaces. One dimer is structurally similar to the four helix-bundle of the classical ferritin monomer9 and is formed by the two main helices of two EncFtn monomers. This dimer contains the di-iron ferroxidase centre (FOC) active site and is known as the FOC dimer (Fig. 1B). Two glutamates (Glu30 and Glu60) and one histidine (His63) per monomer coordinate the iron ions at the interface of the FOC dimer.3 In the pentamer of dimers assembly, FOC dimers are arranged in an annular architecture with a second dimer interface, the non-FOC dimer interface (Fig. 1C) connecting five FOC dimers (Fig. 1C). The non-FOC dimer interface is more extensive and mediated via a mixture of hydrophobic interactions, hydrogen bonds, and salt bridges.9
 | ||
| Fig. 1 (A) Annular pentamer of dimers structure of EncFtn (PDB ID: 5DA5) with transparent solvent accessible surface area (SASA) over cartoon secondary structure. (B) The FOC dimer containing two iron ions bound in the FOC shown with cartoon secondary structure and transparent SASA, and a simplified representation. (C) The non-FOC dimer in cartoon and simplified representation. The two possible assembly pathways of EncFtn via dimer association; the FOC assembly pathway (D) and the non-FOC assembly pathway (E). | ||
Although these crystallographic studies afford us insight into the atomic details of the EncFtn protein structure, these high resolution ‘snapshots’ offer limited insight into the dynamics and assembly pathways of protein complexes. In contrast, structural mass spectrometry methods such as native mass spectrometry (MS) and ion mobility (IM) can report on protein structural/conformational heterogeneity and structural dynamics in a relatively unbiased manner. Consequently, MS-based techniques have proven highly complementary to traditional ‘high-resolution’ structural biology methods.10
We have previously used native MS to demonstrate that removal of the conserved histidine FOC-ligand in the EncFtn from Rhodospirillum rubrum results in loss of the decametric quaternary structure; as a Histidine to Alanine variant of EncFtn exists as only monomer and dimer oligomerisation states.3 In addition, prior native MS studies have also revealed that the EncFtn from the halophile Haliangium Ochraceum exists as a dynamic structure in multiple even-numbered oligomerisation states (dimer, tetramer, hexamer and decamer).9 This observation implies that higher order oligomers are formed by the association of dimer units; presumably either via FOC dimer association, or non-FOC dimer association (Fig. 1D and E).
Herein, we build on these previous findings and employ several complimentary structural proteomic techniques to investigate the dimer association pathway leading to the annular EncFtn structure. We produced both wild type sequence and H63A variants of H. Ochraceum EncFtn (Hoch-WT and Hoch-H63A) as truncated variants (lacking the C-terminal encapsulin localization sequence), by heterologous expressed in Escherichia coli.11 After purification, the masses of both Hoch-WT and Hoch-H63A were verified by LC-MS (Table S2, ESI†).
We first analysed the oligomerisation states of Hoch-WT and Hoch-H63A using native MS (Fig. 2). As previously reported, native MS analysis of Hoch-WT reveals a series of oligomerisation states.9 The major species is dimer (+9 to +11) (Fig. 2A, green circles), with some tetramer (13+ to 15+), hexamer (17+ to 18+) and decamer (22+ to 25+) species also observed (Fig. 2A, purple squares, orange diamonds and pink triangles respectively). Native MS analysis of Hoch-H63A shows that it exists solely as a dimer (7+ to 12+) (Fig. 2A, green circles), demonstrating that the H63A mutation in the H. Ochraceum EncFtn disrupts the formation of higher order EncFtn structures in a similar manner to previously reported for R. rubrum EncFtn.3 This was supported by using non-denaturing PAGE where Hoch-WT appeared in multiple oligomerisation states of varying size; whereas Hoch-H63A was only observed in one oligomerisation state (Fig. S2, ESI†).
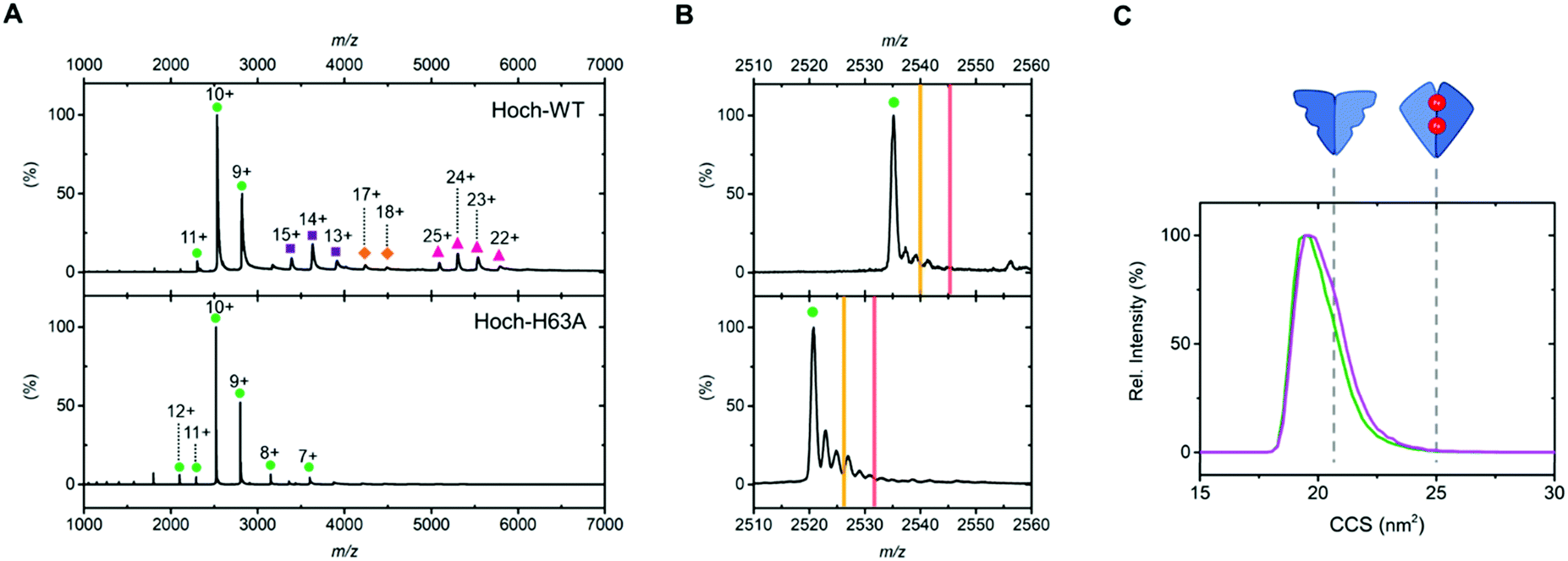 | ||
| Fig. 2 (A) Native nanoelectrospray ionization mass spectra of Hoch-WT and Hoch-H63A with gas phase oligomerization states stressed with coloured shapes. Dimer as green circles; tetramer as purple squares; hexamer as orange diamonds and decamer as pink triangles. (B) 10+ dimer charge states of Hoch-WT (top panel) and Hoch-H63A (bottom panel). Theoretical m/z values of dimers with no metal bound; dimers with one iron bound and two irons bound are highlighted by green circles, yellow lines and red lines respectively. (C) Ion mobility drift time profiles of the 10+ charge states of Hoch-WT (green) and Hoch-H63A (purple). The theoretical collision cross sections of the non-FOC and FOC dimer are shown in dashed grey lines and were calculated using IMPACT software.13 | ||
Having established that dimer assemblies were present in both Hoch-WT and Hoch-H63A, native ion mobility MS was used to investigate their conformation and compare their observed collision cross section (CCS).12 The IM-MS arrival time profiles of the dimer charge states of Hoch-WT and Hoch-H63A were highly similar – after CCS calculation both Hoch-WT and H63A dimers displayed CCS values of ∼19.6 nm2 and presented with similar widths in the CCS profiles (FWHM ∼ 2.1 and 2.3 nm2; Fig. 2C). The similarity of the mobility profiles of the Hoch-WT and Hoch-H63A dimers suggest that they adopt the same dimer form. Moreover, when theoretical CCS values of the two distinct dimer subcomplexes were calculated from the crystal structure of Hoch-WT,13 it is clear that they display significant differences in theoretical CCS. The theoretical CCS of the non-FOC dimer is 21.4 nm2 and for the FOC dimer is 25.3 nm2. Comparison of the experimentally determined CCS values suggests that the dimer form present in both Hoch-WT and Hoch-H63A is the non-FOC dimer (Fig. 2C).
Molecular mass measurement by native MS can also inform on metal content and stoichiometry in metalloproteins. The 10+ charge state of the dimer of Hoch-WT displays several adducts of varying molecular mass; however, none of these adducts are consistent with the coordination of iron (Fe2+: theoretical monoisotopic Δmass +54 Da); and we attribute these to non-specific sodium and potassium adduction (commonly observed in native MS). Similarly, the 10+ Hoch-H63A dimer displays no iron coordination (Fig. 2B). The absence of Fe coordination supports the ion mobility data suggesting non-FOC dimer assembly in both variants (Fig. 1C). The charge states of higher order species of Hoch-WT present as wide peaks, due to overlapping signals of adducted species and the limited mass resolving power at molecular mass >40 kDa. Thus, metal coordination could not be determined for these charge states. Inductively coupled plasma (ICP) MS analysis of Hoch-H63A revealed no substantial iron content (5.6%, Table S4, ESI†). In contrast, significant iron content (albeit at sub-stoichiometric levels) was observed in Hoch-WT (29.1%, Table S4, ESI†), suggesting that iron may be associated with the higher order oligomers of Hoch-WT.
In order to investigate iron-loading in these higher molecular mass species and overcome the mass resolution limitation, Hoch-WT was analysed by native MS on a high-resolution FT-ICR MS platform. This instrument affords isotopic resolution, even at high mass, allowing discrimination of overlapping adduct species and more confident assignment of coordinated metals. This analysis confirmed no iron coordination in the dimer forms of Hoch-WT and Hoch-H63A (Fig. S3, ESI†). FT-ICR MS analysis of the 13+ tetrameric Hoch-WT allowed us to distinguish several adducted forms – which could be assigned as a prominent potassiated series, in addition an isotope distribution corresponding to the coordination of 2Fe2+ (Hoch-WT4Fe22+) was clearly present (Fig. 3). Interestingly, no signal was observed to suggest coordination of a single iron, suggesting the presence of a single di-nuclear FOC centre in the tetrameric species. Taken together these observations match the predicted stoichiometry of the non-FOC assembly pathway (Fig. 1E) i.e. that higher order EncFtn species are assembled from non-FOC dimer association, and that assembly of two non-FOC dimers result in the formation of one di-iron containing FOC.
 | ||
| Fig. 3 (A) High resolution FT-ICR MS analysis of 13+ charge state of the Hoch-WT tetramer. The theoretical isotope distributions of Hoch-WT4 and Hoch-WT4Fe22+ species are highlighted by purple and red circles respectively. Hoch-WT potassiated series shown by blue lines. (B) Region of the spectra which contains overlapping isotope distributions consistent with Hoch-WT4Fe22+ (shown in red) and Hoch-WT4K3+ (shown in blue). | ||
This hypothesis is strengthened by titration of the Hoch-WT with Fe2+ prior to native MS analysis, which resulted in an increased intensity of higher oligomerisation states (tetramer and hexamer in particular) (Fig. S4, ESI†); suggesting that the association of dimers to form higher order subcomplexes is iron dependent.
To complement our native MS experiments, we employed hydrogen deuterium exchange (HDX) MS (see ESI† for further details). By comparing the rate of backbone amide hydrogen exchange in Hoch-WT and Hoch-H63A, we could ascertain regions which differ in solvent exposure and/or dynamics. This analysis highlighted two regions of the Hoch-H63A variant which undergo exchange significantly faster than the wild-type protein – both are located in the central region of the primary sequence and span aa33–42 and aa46–61 (Fig. 4A, peptides ii and iii).
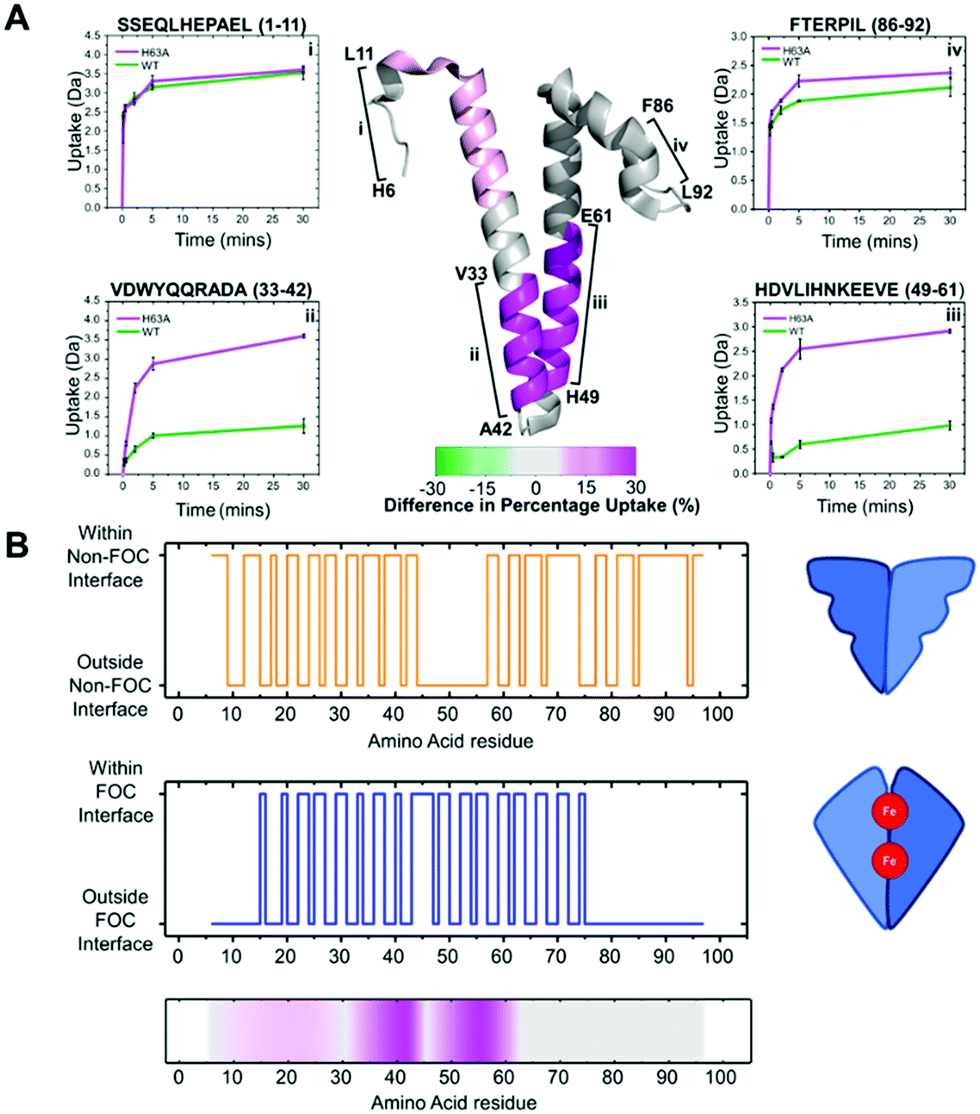 | ||
| Fig. 4 (A) The difference in HD exchange at 30 minutes of D2O labelling mapped on to the monomer crystal structure of Hoch-WT. Regions displaying increased exchange in H63A are coloured pink. HDX uptake plots are shown for four peptides (i–iv) highlighted in the main figure. (B) Structural analysis of Hoch-WT to determine the interface regions for both the FOC (blue) and non-FOC (orange) dimer subcomplexes. The differential HDX data is represented below mapped onto the primary sequence of Hoch-WT (colour scheme used as in (A)). | ||
These regions of the primary sequence, protected from exchange in Hoch-WT, were mapped onto the higher order structure in order to determine their relationship to the two discrete dimer forms of the assembly. First, we delineated the regions of the Hoch-WT protein primary sequence which lie at the interfaces of the two dimers in the decamer assembly using PDBePISA14 to analyse the Hoch-WT structure (PDB 5N5F, Fig. 4B). Although the helical nature of the protein results in partitioning of the side chains at either face of the helical bundles, it was possible to determine regions of the primary sequence associated with each interface. In general, amino acids which reside in the FOC dimer interface are located in the central region of the primary sequence – between aa16–aa75; with extended regions at both the N- and C-termini that do not present in the interface (Fig. 4B, blue). In contrast, the non-FOC interface is more extensive and involves residues at both the N- and C-termini. However, we note a central extended region (aa45–aa57) which is not present at the non-FOC interface (Fig. 4B, orange). In light of this structural interface analysis, it is clear that the regions of the primary sequence protected from exchange in Hoch-WT span regions of the protein that correlate with the FOC–dimer interface (Fig. 4B); most evident from the region spanning aa46–61. This is consistent with the FOC interface being more accessible in Hoch H63A when compared to Hoch-WT.
These findings suggest that Hoch-H63A is present exclusively as the non-FOC dimer; as such, while the non-FOC dimer interface is shielded from exchange, the FOC dimer interface is exposed. In comparison, our native MS analysis reveals that the wild-type protein is present as dimers and higher order assemblies of dimers. Therefore, although it shares the same shielded non-FOC dimer interface as Hoch-H63A, the FOC interface, exposed in Hoch-H63A, will be somewhat shielded by forming these higher order species. Thus, HDX analysis further supports the hypothesis that the predominant dimeric form of Hoch-WT is the non-FOC dimer, and decamer formation is via assembly of these non-FOC dimer subunits.
In conclusion, the solution and gas phase studies of H. Ochraceum EncFtn presented here elucidate the assembly pathway of the characteristic pentamer of dimers arrangement found in EncFtns. The decameric assembly is associated by the addition of the non-FOC dimers via the coordination of iron by FOC residues, including Histidine 63, and the formation of the FOC interface. Our results highlight the dynamic nature of the FOC in the EncFtn proteins and its dual role in mediating the oligomerisation and enzymatic activity of these proteins.
Protein oligomerisation is energetically and evolutionarily favourable and many important classes of enzyme have active sites that are split between subunits in both homo- and hetero-oligomeric structures.15,16 Metal-mediated multimerization is also widespread in metalloproteins.17–20 In this work we have demonstrated the first instance of a ferritin whose assembly is mediated by iron-binding and the formation of the ferroxidase centre between EncFtn monomers. This assembly pathway of the EncFtn decamer, from non-FOC dimers, is distinct to other members of the ferritin family. While the FOC interface closely resembles the four-helix bundle FOC of the classical and bacterioferritins; the role of the FOC in stabilising the functional quaternary structure is unique to the EncFtn class of the ferritin-like proteins.
This work was financially supported by the BBSRC (Grant Number BB/N005570/1 and BB/R013993/1). J. R. is funded via the BBSRC EastBIO doctoral training programme (BB/M010996/1). T. L. is funded via an EPSRC PhD studentship and the School of Chemistry, UoE.
Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Bou-Abdallah, Biochim. Biophys. Acta, Gen. Subj., 2010, 1800, 719–731 CrossRef CAS PubMed.
- X. Liu and E. C. Theil, Acc. Chem. Res., 2005, 38, 167–175 CrossRef CAS PubMed.
- D. He, S. Hughes, S. Vanden-Hehir, A. Georgiev, K. Altenbach, E. Tarrant, C. L. Mackay, K. J. Waldron, D. J. Clarke and J. Marles-Wright, eLife, 2016, 5, e18972 CrossRef PubMed.
- C. A. McHugh, J. Fontana, D. Nemecek, N. Cheng, A. A. Aksyuk, J. B. Heymann, D. C. Winkler, A. S. Lam, J. S. Wall, A. C. Steven and E. Hoiczyk, EMBO J., 2014, 33, 1896–1911 CrossRef CAS PubMed.
- F. Akita, K. T. Chong, H. Tanaka, E. Yamashita, N. Miyazaki, Y. Nakaishi, M. Suzuki, K. Namba, Y. Ono, T. Tsukihara and A. Nakagawa, J. Mol. Biol., 2007, 368, 1469–1483 CrossRef CAS PubMed.
- T. W. Giessen and P. A. Silver, Nat. Microbiol., 2017, 2, 17029 CrossRef PubMed.
- M. Sutter, D. Boehringer, S. Gutmann, S. Günther, D. Prangishvili, M. J. Loessner, K. O. Stetter, E. Weber-Ban, N. Ban, A. Steven, E. Hoiczyk, N. Moriarty, R. Oeffner, R. Read, D. Richardson, J. Richardson, T. Terwilliger and P. Zwart, Nat. Struct. Mol. Biol., 2008, 15, 939–947 CrossRef CAS PubMed.
- W. F. Rurup, J. Snijder, M. S. T. Koay, A. J. R. Heck and J. J. L. M. Cornelissen, J. Am. Chem. Soc., 2014, 136, 3828–3832 CrossRef CAS PubMed.
- D. He, C. Piergentili, J. Ross, E. Tarrant, L. R. Tuck, C. Logan Mackay, Z. McIver, K. J. Waldron, D. J. Clarke and J. Marles-Wright, Biochem. J., 2019, 476, 975–989 CrossRef CAS PubMed.
- A. J. R. Heck, Nat. Methods, 2008, 5, 927–933 CrossRef CAS PubMed.
- S. V. Iverson, T. L. Haddock, J. Beal and D. M. Densmore, ACS Synth. Biol., 2016, 5, 99–103 CrossRef CAS PubMed.
- E. Jurneczko and P. E. Barran, Analyst, 2011, 136, 20–28 RSC.
- E. G. Marklund, M. T. Degiacomi, C. V. Robinson, A. J. Baldwin and J. L. P. Benesch, Structure, 2015, 23, 791–799 CrossRef CAS PubMed.
- E. Krissinel and K. Henrick, J. Mol. Biol., 2007, 372, 774–797 CrossRef CAS PubMed.
- H. J. Chen, T. P. Ko, C. Y. Lee, N. C. Wang and A. H. J. Wang, Structure, 2009, 17, 517–529 CrossRef CAS PubMed.
- K. R. Malley, O. Koroleva, I. Miller, R. Sanishvili, C. M. Jenkins, R. W. Gross and S. Korolev, Nat. Commun., 2018, 9, 765 CrossRef PubMed.
- A. C. Partin, T. D. Ngo, E. Herrell, B. C. Jeong, G. Hon and Y. Nam, Nat. Commun., 2017, 8, 1737 CrossRef PubMed.
- M. M. He, S. L. Clugston, J. F. Honek and B. W. Matthews, Biochemistry, 2000, 39, 8719–8727 CrossRef CAS PubMed.
- M. A. Kihlken, A. P. Leech and N. E. Le Brun, Biochem. J., 2002, 368, 729–739 CrossRef CAS PubMed.
- R. Dutoit, T. Van Gompel, N. Brandt, D. Van Elder, J. Van Dyck, F. Sobott and L. Droogmans, J. Biol. Chem., 2019, 294, 17777–17789 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc08130e |
| This journal is © The Royal Society of Chemistry 2020 |