
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biotinylated photoactive Pt(IV) anticancer complexes†
Huayun
Shi
 ,
Cinzia
Imberti
,
Huaiyi
Huang
,
Ian
Hands-Portman
and
Peter J.
Sadler
*
,
Cinzia
Imberti
,
Huaiyi
Huang
,
Ian
Hands-Portman
and
Peter J.
Sadler
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: p.j.sadler@warwick.ac.uk
First published on 28th January 2020
Abstract
Novel biotinylated diazido-Pt(IV) complexes exhibit high visible light photocytotoxicity while being stable in the dark. Photocytotoxicity and cellular accumulation of all-trans-[Pt(py)2(N3)2(biotin)(OH)] (2a) were enhanced significantly when bound to avidin; irradiation induced dramatic cellular morphological changes in human ovarian cancer cells treated with 2a.
Tumour-targeting drugs can improve selective drug delivery to cancer cells and reduce undesirable side effects.1–4 Biotin (vitamin H or B7) plays an important role as cellular growth promoter and co-enzyme for carboxylase enzymes.5 Exogenous biotin is taken up via a high-affinity biotin transporter and a sodium-dependent multivitamin transporter (SMVT).6,7 Notably, cancer cells overexpress SMVT, thus making biotin a tumour-targeting vector.8–10 Biotin possesses a high affinity specific for tetrameric egg white avidin through noncovalent bonding with Kd values of 10−13 to 10−15 M.11 The avidin–biotin complex induces a relatively low immune response in vivo and has been widely used in cancer-targeted therapy.5,12–15 Avidin displays strong adherence to cells in vitro, and can be administered as a nano-carrier for delivery of biotinylated drugs.16 Biotinylation of drugs is therefore a feasible strategy to improve their cancer-cell accumulation and selectivity,17,18 and has also been applied in the design of photoactive drugs.19–22
Photoactive Pt agents provide spatial control of cytotoxicity with novel mechanisms of action and lack of cross-resistance with cisplatin.23,24 Diazido Pt(IV) complexes are potent photoactive prodrugs with high dark stability due to their kinetic inertness,25–27 and photocytotoxicity exerted by the combined effects of Pt(II) products, azidyl radicals and reactive oxygen species (ROS) formed on photoactivation.28–30Trans,trans,trans-[Pt(N3)2(OH)2(py)2] (1) kills cancer cells upon irradiation with high photocytotoxicity (dark versus light) indices.27 Axial OH ligands enhance aqueous solubility of Pt(IV) complexes and stabilise the PtIV oxidation state.31 Derivatisation of the releasable axial ligand can improve the pharmacological properties without altering the mechanism of action.32–34
Herein, we have prepared two novel biotinylated diazido Pt(IV) complexes trans,trans,trans-[Pt(py)2(N3)2(biotin)(OH)] (2a) and trans,trans,trans-[Pt(py)2(N3)2(biotin)(DCA)] (2b, Scheme 1). The pyruvate dehydrogenase kinase (PDK) inhibitor dichloroacetate (DCA) can be delivered to cancer cells effectively as an axial ligand in Pt(IV) prodrugs.35–37 Dark stability, photodecomposition, photoreactions with 5′-GMP and DNA, and interaction with avidin were investigated for 2a and 2b, and their photocytotoxicity and cellular accumulation compared to those of avidin-complex adducts. Cellular morphological changes upon treatment of cancer cells with 2a were examined by confocal microscopy.
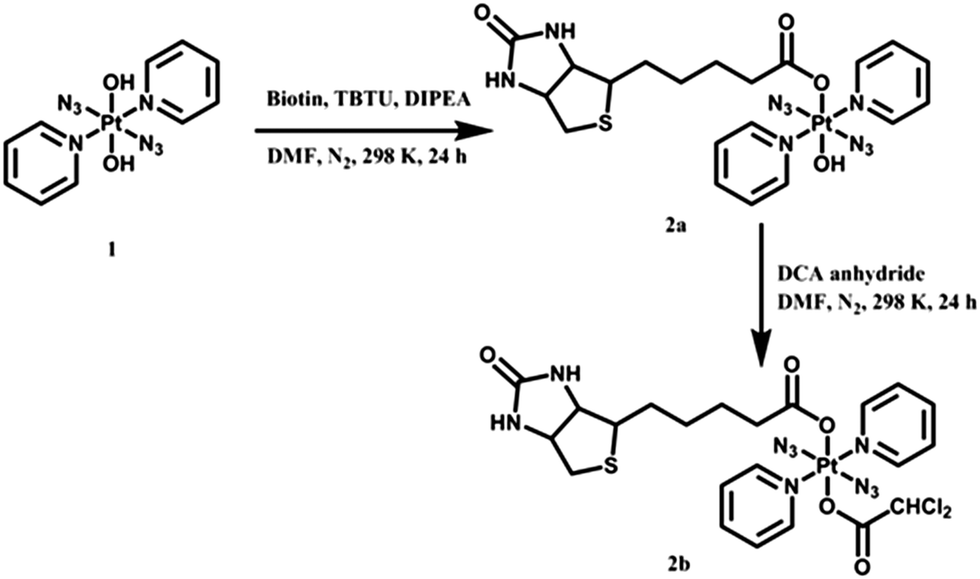 | ||
| Scheme 1 Synthetic routes for biotinylated complexes 2a and 2b. | ||
The synthetic routes for photoactive Pt(IV) complexes 2a and 2b are summarised in Scheme 1. Mono-substituted 2a was obtained by combining 1 with biotin using TBTU/DIPEA as coupling agents, while the bi-substituted 2b was synthesised by stirring 2a with DCA anhydride. Both complexes gave satisfactory elemental analyses and were also characterised by ESI-HRMS, NMR and UV-vis spectroscopy, and for purity by HPLC (Fig. S1, ESI†).
The 1H and 13C NMR data are consistent with the proposed structures of the complexes. The Pt-coordinated pyridine has characteristic 1H NMR doublets with 195Pt satellites at ca. 8.8 ppm, and the triplets at ca. 8. 3 and 7.9 ppm, with 13C NMR resonances at ca. 150, 143 and 127 ppm (Fig. S2–S5, ESI†). Two typical singlets at 6.38 and 6.35 ppm assignable to NHC(O)NH of biotin were detected for both complexes. The electronic absorption spectra of 2a and 2b in aqueous solution were similar, although 2b exhibited a slight red shift of the LMCT (N3 → Pt) transition and stronger absorption at wavelengths <270 nm due to the presence of the DCA ligand.
Complexes 2a and 2b exhibited excellent dark stability in RPMI-1640/DMSO (95%/5%, v/v) monitored by UV-vis spectroscopy (Fig. 1a and Fig. S6a, ESI†). When irradiated with blue light (420 nm), the LMCT bands of both complexes at ca. 300 nm decreased in intensity, indicating release of the azide ligands (Fig. 1b and Fig. S6b, ESI†).27 Notably, 2b exhibited a larger decrease at the absorption maximum, probably due to release of the DCA ligand. Released azidyl radicals and singlet oxygen were trapped by DMPO and TEMP, respectively, for 2a upon irradiation (463 nm; Fig. 1c and d).
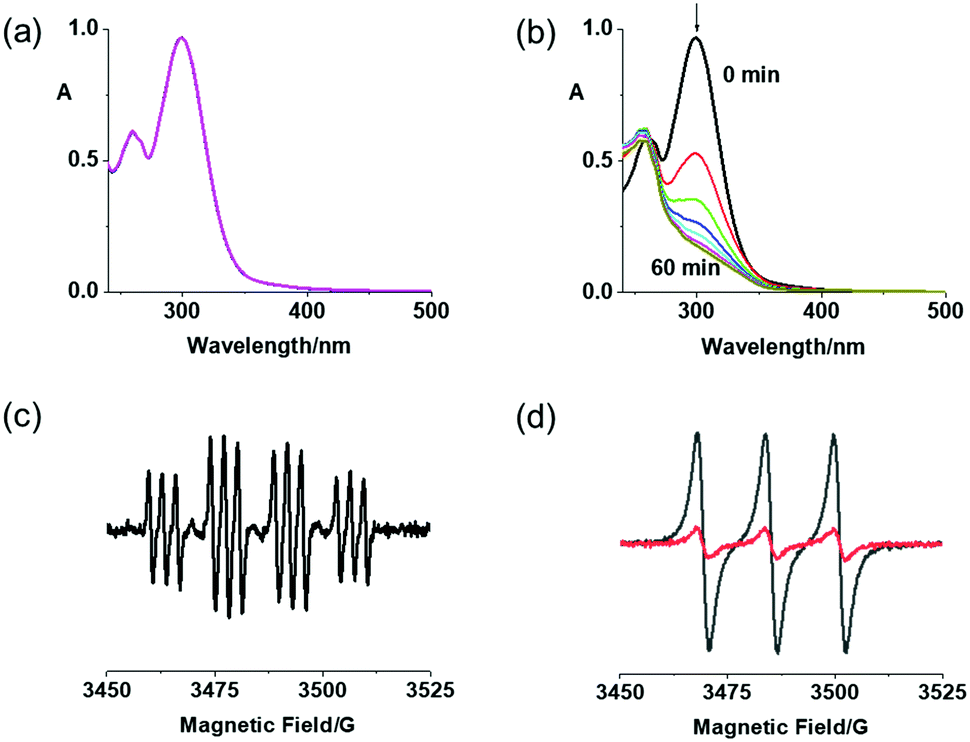 | ||
Fig. 1 (a) UV-vis spectral changes for 2a (50 μM) in RPMI-1640 in the dark for 2 h, and (b) in H2O upon 1 h irradiation with blue light (420 nm); (c) EPR spectra for 2a (2.5 mM) with DMPO in H2O after irradiation (463 nm), and (d) with TEMP in CH3CN in the dark ( ) and after irradiation (463 nm, —). ) and after irradiation (463 nm, —). | ||
The photoreactions of 2a/2b with 5′-GMP were analysed by LC-MS. Pt(IV) complexes (30 μM) were incubated with 5′-GMP (2 mol equiv.) at 310 K in the dark for 1 h, then irradiated by blue light (420 nm) for 1 h (Fig. S7, ESI†). For both complexes, [PtII(CH3CN)(py)2(GMP-H)]+ (G1, 756.48 m/z) was detected as the major Pt-GMP product, together with two minor adducts [PtII(HCOO)(py)2(GMP)]+ (G2, 762.18 m/z) and [PtII(N3)(py)2(GMP)]+ (G3, 758.23 m/z). These results were similar to those obtained for 1, suggesting a negligible effect of axial substituents on the photoreaction with guanine.38
DNA melting experiments monitored by UV-vis were carried out in phosphate buffer (1 mM, pH = 7.9) to investigate DNA binding with a drug/base pair ratio of 0.2 (Table S1, ESI†). Similar to parent complex 1, 2a and 2b interact with ct-DNA only weakly (ΔTm < 2 K) in the dark, while upon irradiation with blue light (420 nm) an apparent interaction was observed (ΔTmca. 5 K). The increase in the DNA melting temperature suggests that 2a and 2b might form inter-strand cross-links after irradiation, similar to 1.
The binding affinity of avidin towards 2a and 2b in PBS was assessed by a displacement titration using HABA (2-(4-hydroxyphenylazo)benzoic acid) and monitored by UV-vis spectroscopy (Fig. 2). The avidin–HABA adduct displays an absorption band at ca. 496 nm, which decreases when HABA is replaced by biotin.39 On mixing, a gradual decrease of the absorbance at 496 nm was observed upon addition of either complex to a solution containing HABA (160 μM) and avidin (8 μM), indicating their stronger binding to avidin compared with HABA (dissociation constant Kd = 10−6 M).39 A sharp end point and same absorbance changes (ΔA = 1.01) with biotin were observed for 2a and 2b, indicating their similar affinity towards avidin as unmodified biotin. These results suggested the possibility of using avidin as a nanocarrier for delivery of biotinylated complexes to cancer cells.
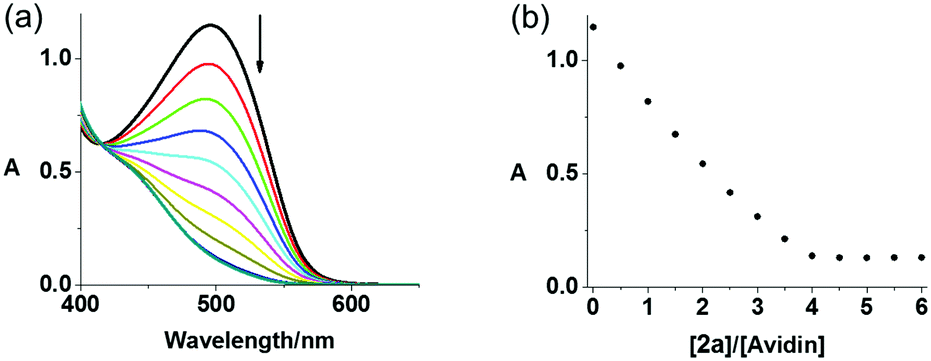 | ||
| Fig. 2 (a) UV-vis spectral changes for avidin–HABA in PBS solution upon addition of complex 2a in DMSO, (b) absorbance change at 496 nm in HABA displacement titrations of biotinylated complexes 2a. | ||
The IC50 values of 2a and 2b in human A2780 ovarian, A549 lung and PC3 prostate cancer cell lines were determined by the sulforhodamine B (SRB) colorimetric assay (Table 1), using parent complex 1 and CDDP (cisplatin) as references. Both complexes were relatively non-toxic towards all cancer cells and healthy MRC-5 lung fibroblasts in the dark with IC50 values >50/100 μM, but exhibited promising photocytotoxicity with high photocytotoxicity indices (PI). In all the cancer cell lines studied, di-substituted 2b (1.3–5.9 μM) is at least twice as phototoxic as the mono-substituted 2a (11.7–21.1 μM), and 5× more toxic than 1 (7.1–55.6 μM). Under the same conditions, cisplatin exhibited very low cytotoxicity in all cell lines (IC50 > 100 μM) due to the short incubation time (2 h).
| Comp. | Cell line/IC50 (μM)a | ||||
|---|---|---|---|---|---|
| A2780 | A549 | PC3 | MRC5 | ||
| a Data are from three independent experiments. | |||||
| 2a | Dark | >100 | >100 | >100 | >100 |
| Irrad | 11.7 ± 0.3 | 13.3 ± 0.7 | 21.1 ± 0.4 | ||
| PI | >8.5 | >7.5 | >4.7 | ||
2a–avi (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
Dark | >100 | |||
| Irrad | 4.4 ± 0.3 | ||||
| PI | >22.7 | ||||
| 2b | Dark | >50 | >100 | >100 | >100 |
| Irrad | 1.3 ± 0.2 | 5.9 ± 0.6 | 3.0 ± 0.1 | ||
| PI | >38.5 | >16.9 | >33.3 | ||
|
2b–avi (4:1) |
Dark | >50 | |||
| Irrad | 2.45 ± 0.01 | ||||
| PI | >20.4 | ||||
| 1 | Dark | >100 | >100 | >100 | >100 |
| Irrad | 7.1 ± 0.4 | 51.9 ± 2.5 | 55.6 ± 0.9 | ||
| PI | >14.1 | >1.9 | >1.8 | ||
| CDDP | Dark | >100 | >100 | >100 | >100 |
| Irrad | >100 | >100 | >100 | ||
In A2780 ovarian cancer cells, 2a (IC50 = 11.7 μM) was slightly less active than 1 (IC50 = 7.1 μM), while 2b was more potent (IC50 = 1.3 μM), potentially attributable to the synergistic anticancer activity of the released DCA ligand. In order to investigate the effect of avidin on activity, 2a was mixed with avidin (2a:avidin = 4:1) prior to addition to A2780 cells following the same protocol used for 2a alone. Notably, the avidin–2a complex exhibited good dark stability, being potently photocytotoxic in A2780 cells with an IC50 value of 4.4 μM, 2.7× lower than that of 2a, and 1.6× more toxic than 1. In contrast, the photocytotoxicity of 2b was not enhanced in the presence of avidin.
Since biotin is expected to exhibit a preference towards cancer cells due to overexpressed receptors,5 ICP-MS was used to quantify and compare the cellular uptake of biotinylated complexes 2a and 2b as well as the unsubstituted 1. A2780 ovarian cancer cells were treated with 10 μM prodrugs in the dark for 1 h, then the Pt content of cells was analysed by ICP-MS (Table 2). The mono-substituted 2a exhibited lower Pt accumulation (0.4 ng per 106 cells) than 1 (0.64 ng per 106 cells) after 1 h incubation, while accumulation of di-substituted 2b (21.1 ng per 106 cells) was 33× higher than that of 1 and 53× higher than 2a. These results showed that conjugation with biotin alone did not increase the accumulation of diazido Pt(IV) complexes in A2780 cells, while the substitution of the second axial ligand with DCA resulted in significant increase in Pt cellular accumulation, perhaps due to higher lipophilicity of 2b, consistent with its longer HPLC retention time (12.3 min for 2a, 19.7 min for 2b, Fig. S1, ESI†). Notably, the amount of Pt taken up by A2780 cells incubated with 1, 2a and 2b was inversely proportional to their IC50 values. These results suggested that cellular accumulation of Pt(IV) prodrugs plays an important role in their antiproliferative activity. Since the avidin–2a complex exhibited improved photocytotoxicity, the effect of avidin on the uptake of 2a was also investigated. The accumulation of Pt after exposure of A2780 cells to the mixture of 10 μM 2a and avidin (4:1) was 10× higher than 2a alone, which correlates with the higher photocytotoxicity of 2a–avidin complex. However, Pt accumulation for 1 and 2b was not affected by avidin, since 1 does not contain biotin and 2b is mainly taken up by passive diffusion due to its high lipophilicity.
| Pt accumulation (ng per 106 cells)a | |||
|---|---|---|---|
| Complex | Complex with avidin | ||
| a All experiments were conducted in triplicate. Comparison with untreated controls was performed by a two-tail t-test with unequal variances. *p < 0.05, **p < 0.01, ***p < 0.005. | |||
| 2a | 0.40 ± 0.15* |
2a–avi (4:1) |
4.1 ± 0.7* |
| 2b | 21.1 ± 2.0*** |
2b–avi (4:1) |
20.0 ± 1.5*** |
| 1 | 0.64 ± 0.07*** |
1–avi (4:1) |
0.56 ± 0.09** |
A2780 ovarian cancer cells were treated with 2a (1 or 2× photo IC50 concentration) in the presence and absence of light and live-imaged using confocal microscopy and flow cytometry to investigate changes in cell morphology (Fig. 3 and Fig. S8, S10, ESI†). The cell permeant dye SYTO™ 17 was used to stain the nuclei. Without irradiation, 2a exhibited low cytotoxicity (IC50 > 100 μM) and, accordingly, no changes in cellular morphology were observed when cells were treated with 2a for 2 h in the dark. In these conditions, the cells appeared healthy with well-defined plasma membranes and intact nuclei. In contrast, A2780 cells treated with 2a exhibited dramatic morphological changes after 1 h irradiation with blue light (465 nm). The cells rounded up and the nuclei were fragmented into pieces. Damaged membranes and copious cell debris were observed when cells were treated with 2a at 2 × IC50 concentration (Fig. 3 and Fig. S10, ESI†). DNA is usually regarded as the major target of platinum anticancer drugs, so the ability of 2a to fragment cell nuclei exclusively upon irradiation, indicated its potential as a photoactive prodrug with a novel mechanism of action. Complex 2a at higher concentration induced more morphological changes upon irradiation, while irradiation alone did not result in significant effects on cell morphology (Fig. S9 and S10, ESI†).
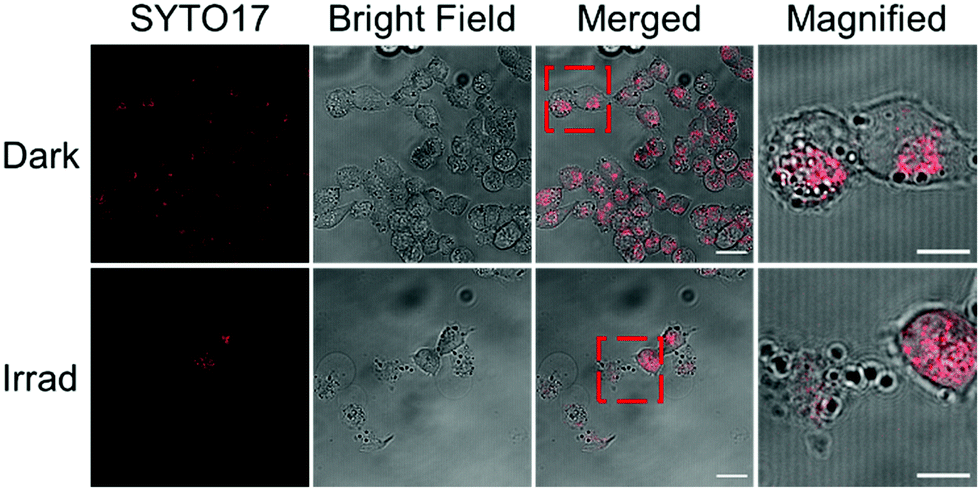 | ||
| Fig. 3 Fluorescence images of A2780 ovarian cancer cells treated with 2a (2 × photo IC50 concentration) in the dark and after irradiation with blue light (λ = 465 nm). Cells were stained by SYTO™ 17 (2.5 μM, λex/λem = 633/638–759 nm) for 30 min. Scale bar = 20 μm (10 μm in magnification). Fragmented nuclei and damaged membranes of A2780 cells treated with 2a were observed upon irradiation, while in the dark, cells were intact and well-defined. | ||
Cellular ROS generation for 2a, 2b and 1 was monitored in A549 lung cancer cells by DCFH-DA, which exhibits switch-on fluorescence in the presence of ROS. Cells treated with complexes (10 μM) in the dark showed no apparent change, but 2b induced an increased DCF fluorescence upon irradiation, indicating its ability to generate ROS (Table S2, ESI†).
In summary, two novel biotinylated Pt(IV) complexes trans,trans,trans-[Pt(py)2(N3)2(biotin)(OH)] (2a) and trans,trans,trans-[Pt(py)2(N3)2(biotin)(DCA)] (2b) have been synthesised and characterised. They exhibited good dark stability and promising photoactivity, releasing azidyl radicals and generating singlet oxygen. Photoreactions with 5′-GMP resulted in the formation of [PtII(CH3CN)(py)2(GMP-H)]+ as a major product. An increase in ct-DNA melting temperature suggested formation of inter-strand crosslinks. Promising photocytotoxicity of 2a and 2b towards human A2780 ovarian, A549 lung and PC3 prostate cancer cells was observed upon irradiation with low-dose blue light (465 nm, 4.8 mW cm−2, 1 h), with high photocytotoxicity indices. The di-substituted 2b exhibited much higher photocytotoxicity and cellular Pt accumulation, compared with mono-substituted 2a and unsubstituted 1. The strong interaction of 2a and 2b with avidin was confirmed by HABA displacement from avidin–HABA. When A2780 ovarian cancer cells were incubated with 1:4 avidin:2a, both photocytotoxicity and cellular accumulation increased significantly. Dramatic morphological changes in A2780 cells were observed after irradiation with 2a, consistent with its promising photocytotoxicity.
We acknowledge financial support from the EPSRC (EP/G006792, EP/F034210/1 to PJS), University of Warwick (Chancellor's International PhD Scholarship for HS), Royal Society (grant no. NF160307, Newton International Fellowship for HH), and Wellcome Trust (grant no. 209173/Z/17/Z, Si Henry Wellcome Fellowship for CI).
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. L. Ma, D. S. H. Chan and C. H. Leung, Chem. Soc. Rev., 2013, 42, 2130–2141 RSC.
- L. Cai, Z. Gu, J. Zhong, D. Wen, G. Chen, L. He, J. Wu and Z. Gu, Drug Discovery Today, 2018, 23, 1126–1138 CrossRef CAS PubMed.
- H. Ijaz, J. Qureshi, U. R. Tulain, F. Iqbal, Z. Danish, A. Fayyaz and A. Sethi, Bioinspired, Biomimetic Nanobiomater., 2018, 7, 109–121 CrossRef.
- E. R. Ruskowitz and C. A. DeForest, Nat. Rev. Mater., 2018, 17087 CrossRef CAS.
- W. X. Ren, J. Han, S. Uhm, Y. J. Jang, C. Kang, J. H. Kim and J. S. Kim, Chem. Commun., 2015, 51, 10403–10418 RSC.
- J. Zempleni and D. M. Mock, Am. J. Physiol., 1998, 275, C382–C388 CrossRef CAS PubMed.
- K. Balamurugan, A. Ortiz and H. M. Said, Am. J. Physiol., 2003, 285, G73–G77 CAS.
- P. D. Prasad, H. Wang, W. Huang, Y. J. Fei, F. H. Leibach, L. D. Devoe and V. Ganapathy, Arch. Biochem. Biophys., 1999, 366, 95–106 CrossRef CAS PubMed.
- S. Luo, V. S. Kansara, X. Zhu, D. Pal and A. K. Mitra, Mol. Pharming, 2006, 3, 329–339 CrossRef CAS PubMed.
- H. Wang, W. Huang, W. Fei, H. Xia, T. L. Y. Feng, F. H. Leibac, L. D. Devoe, V. Ganapathy and P. D. Prasad, J. Biol. Chem., 1999, 274, 14875–14883 CrossRef CAS PubMed.
- N. M. Green, Adv. Protein Chem., 1975, 29, 85–133 CrossRef CAS PubMed.
- H. P. Lesch, M. U. Kaikkonen, J. T. Pikkarainen and S. Ylä-Herttuala, Expert Opin. Drug Delivery, 2010, 7, 551–564 CrossRef CAS PubMed.
- X. Zeng, Y. Sun, X. Zhang and R. Zhuo, Org. Biomol. Chem., 2009, 7, 4201–4210 RSC.
- A. Jain and K. Cheng, J. Controlled Release, 2017, 245, 27–40 CrossRef CAS PubMed.
- S. C. Park, Y. M. Kim, N. H. Kim, E. J. Kim, Y. H. Park, J. R. Lee and M. K. Jang, Macromol. Res., 2017, 25, 882–889 CrossRef CAS.
- L. Chen, B. Schechter, R. Arnon and M. Wilchek, Drug Dev. Res., 2000, 50, 258–271 CrossRef CAS.
- N. Muhammad, N. Sadia, C. Zhu, C. Luo, Z. Guo and X. Wang, Chem. Commun., 2017, 53, 9971–9974 RSC.
- S. Maiti, N. Park, J. H. Han, H. M. Jeon, J. H. Lee, S. Bhuniya, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 4567–4572 CrossRef CAS PubMed.
- K. Li, L. Qiu, Q. Liu., G. Lv, X. Zhao, S. Wang and J. Lin, J. Photochem. Photobiol., B, 2017, 174, 243–250 CrossRef CAS PubMed.
- B. Siewert, M. Langerman, A. Pannwitz and S. Bonnet, Eur. J. Inorg. Chem., 2018, 4117–4124 CrossRef CAS PubMed.
- J. Li, L. Zeng, K. Xiong, T. W. Rees, C. Jin, W. Wu, Y. Chen, L. Jia and H. Chao, Chem. Commun., 2019, 55, 10972–10975 RSC.
- B. Ghazal, E. N. Kaya, A. Husain, A. Ganesan, M. Durmuş, S. Makhseed and J. Porphyr, Phthalocyanines, 2019, 23, 46–55 CrossRef CAS.
- S. Bonnet, Dalton Trans., 2018, 47, 10330–10343 RSC.
- A. Bjelosevic, B. J. Pages, L. K. Spare, K. M. Deo, D. L. Ang and J. R. Aldrich-Wright, Curr. Med. Chem., 2018, 25, 478–492 CrossRef CAS PubMed.
- P. Müller, B. Schröder, J. A. Parkinson, N. A. Kratochwil, R. A. Coxall, A. Parkin, S. Parsons and P. J. Sadler, Angew. Chem., Int. Ed., 2003, 42, 335–339 CrossRef PubMed.
- F. S. Mackay, J. A. Woods, P. Heringová, J. Kašpárková, A. M. Pizarro, S. A. Moggach, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20743–20748 CrossRef CAS PubMed.
- N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. Mackay and P. J. Sadler, Angew. Chem., Int. Ed., 2010, 49, 8905–8908 CrossRef CAS PubMed.
- Y. Zhao, N. J. Farrer, H. Li, J. S. Butler, R. J. McQuitty, A. Habtemariam, F. Wang and P. J. Sadler, Angew. Chem., Int. Ed., 2013, 52, 13633–13637 CrossRef CAS PubMed.
- J. S. Butler, J. A. Woods, N. J. Farrer, M. E. Newton and P. J. Sadler, J. Am. Chem. Soc., 2012, 134, 16508–16511 CrossRef CAS PubMed.
- J. Pracharova, L. Zerzankova, J. Stepankova, O. Novakova, N. J. Farrer, P. J. Sadler, V. Brabec and J. Kasparkova, Chem. Res. Toxicol., 2012, 25, 1099–1111 Search PubMed.
- M. D. Hall and T. W. Hambley, Coord. Chem. Rev., 2002, 232, 49–67 CrossRef CAS.
- A. Gandioso, E. Shaili, A. Massaguer, G. Artigas, A. González-Cantó, J. A. Woods, P. J. Sadler and V. Marchán, Chem. Commun., 2015, 51, 9169–9172 RSC.
- E. Shaili, M. Fernández-Giménez, S. Rodríguez-Astor, A. Gandioso, L. Sandín, C. García-Vélez, A. Massaguer, G. J. Clarkson, J. A. Woods, P. J. Sadler and V. Marchán, Chem. – Eur. J., 2015, 21, 18474–18486 CrossRef CAS PubMed.
- H. Shi, Q. Wang, V. Venkatesh, G. Feng, L. S. Young, I. Romero-Canelón, M. Zeng and P. J. Sadler, Dalton Trans., 2019, 48, 8560–8564 RSC.
- E. Wexselblatt, E. Yavin and D. Gibson, Angew. Chem., Int. Ed., 2013, 52, 6059–6062 CrossRef CAS PubMed.
- S. Dhara and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22199–22204 CrossRef PubMed.
- E. Petruzzella, J. P. Braude, J. R. Aldrich-Wright, V. Gandin and D. Gibson, Angew. Chem., Int. Ed., 2017, 56, 11539–11544 CrossRef CAS PubMed.
- R. R. Vernooij, T. Joshi, M. D. Horbury, B. Graham, E. I. Izgorodina, V. G. Stavros, P. J. Sadler, L. Spiccia and B. R. Wood, Chem. – Eur. J., 2018, 24, 5790–5803 CrossRef CAS PubMed.
- N. M. Green, Biochem. J., 1965, 94, 23c–24c CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, photodecomposition, UV-vis titration, and experimental details. See DOI: 10.1039/c9cc07845b |
| This journal is © The Royal Society of Chemistry 2020 |