
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNucleoside-modified AdoMet analogues for differential methyltransferase targeting†
Nicolas V.
Cornelissen
 ,
Freideriki
Michailidou
,
Fabian
Muttach
,
Kristina
Rau
and
Andrea
Rentmeister
*
,
Freideriki
Michailidou
,
Fabian
Muttach
,
Kristina
Rau
and
Andrea
Rentmeister
*
Department of Chemistry, Institute of Biochemistry, University of Muenster, Wilhelm-Klemm-Straße 2, D-48149 Muenster, Germany. E-mail: a.rentmeister@uni-muenster.de
First published on 22nd January 2020
Abstract
Methyltransferases (MTases) modify a wide range of biomolecules using S-adenosyl-L-methionine (AdoMet) as the cosubstrate. Synthetic AdoMet analogues are powerful tools to site-specifically introduce a variety of functional groups and exhibit potential to be converted only by distinct MTases. Extending the size of the substituent at the sulfur/selenium atom provides selectivity among MTases but is insufficient to discriminate between promiscuous MTases. We present a panel of AdoMet analogues differing in the nucleoside moiety (NM-AdoMets). These NM-AdoMets were efficiently produced by a previously uncharacterized methionine adenosyltransferase (MAT) from methionine and ATP analogues, such as ITP and N6-propargyl-ATP. The N6-modification changed the relative activity of three representative MTases up to 13-fold resulting in discrimination of substrates for the methyl transfer and could also be combined with transfer of allyl and propargyl groups.
Methyltransferases (MTases) modify a plethora of biomolecules, including small molecules, DNA, RNA and histones by site-specific introduction of a methyl group originating most often from S-adenosyl-L-methionine (AdoMet or SAM).1 The function of secondary metabolites and biological processes like regulation in epigenetics and epitranscriptomics critically depend on precisely positioned methyl groups and – in some cases – their timed removal.2–5
In combination with a non-natural AdoMet analogue, many MTases can also transfer longer alkyl chains, rendering them invaluable for site-specific late-stage alkylation, alkyl randomization or for precise modification of nucleic acids and histones.6–13 The post-synthetic enzymatic modification together with bioorthogonal chemistry has become a powerful tool to label complex biomolecules at chemically indistinguishable sites, but also to purify them from the cell lysate or to photo-crosslink interaction partners.14–20 Even effects on the biological function and reversibility could be demonstrated.21,22
Importantly, AdoMet can be generated enzymatically from methionine and ATP using methionine adenosyl transferase (MAT), also referred to as SAM synthetase, an enzyme present in all organisms. We and others reported on the substrate promiscuity of different MAT enzymes and showed that numerous methionine analogues can be used by different WT and engineered MATs in combination with a suitable MTase to prevent product inhibition.23–27 Human MAT2a (hMAT2a) and its variant I117A proved particularly promiscuous with regard to substituents at the sulfur atom.28 This provides the groundwork for novel metabolic labeling approaches based on methionine analogues that enabled enrichment of MTase target sites in RNAs and histones, circumventing the need to bring cell-impermeable AdoMet analogues that have limited stability in aqueous solution into the cell.28,29
However, there are more than 300 MTases encoded in the human genome according to the UniProt database.31 While the idea is tempting to target select MTases by engineering a particular MTase for accepting AdoMet analogues with large substituents at the sulfur atom, recent evidence indicates that a fraction of MTases is extremely promiscuous and does not face size limitations, because the substituent can reach out of the binding cleft.20,21,32,33
We therefore think that MTases cannot be selectively targeted by merely extending the size of the substituent. Instead, we hypothesized that AdoMet analogues bearing modifications at the adenosine moiety in addition to the sulfur/selenium atom might be a better way to target certain MTases, reminiscent of kinase targeting approaches34,35 (Fig. 1). This idea is supported by work in the field of MTase inhibitors, where S-adenosylhomocysteine (SAH) analogues with modifications at the N6-position of the adenine ring proved favorable and could even be combined with a “bump-and-hole” approach.36,37 The use of such AdoMet analogues would be attractive to achieve selectivity in metabolic labeling approaches and make biomolecules accessible to analysis after cell harvesting. Recently, enzymatic in situ generation from 5′-chloro-5′-deoxyadenosine analogues was shown,38 but formation from ATP analogues and methionine has not been explored to the best of our knowledge.
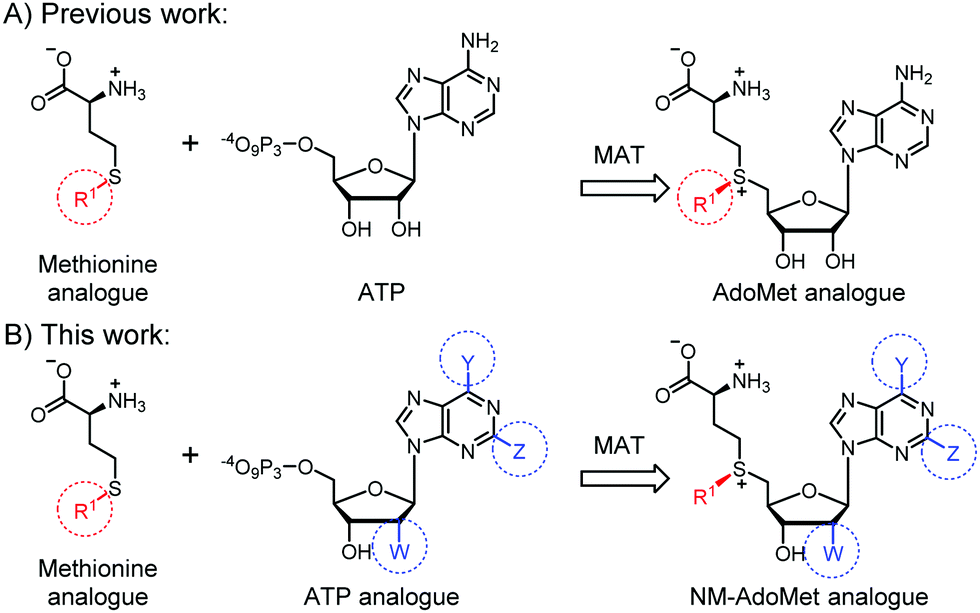 | ||
| Fig. 1 (A) Previous work showed that methionine analogues and ATP can be converted by methionine adenosyltransferase (MAT) to AdoMet analogues.23–26,30 These were used as cosubstrates in alkylation reactions catalyzed by promiscuous methyltransferases (MTases). (B) In this work, we show that methionine or analogues together with ATP analogues can be converted to nucleoside-modified AdoMet analogues (NM-AdoMets) allowing for selectivity among different MTases. | ||
We therefore sought to explore the promiscuity of MATs regarding the adenosine moiety of ATP and used human MAT2a variant I117A (hMAT2a-Var) that was previously reported to be rather promiscuous.39 In addition, we tested the putative wildtype MAT from Cryptosporidium hominis (ChMAT) that had been crystallized by the Seattle Structural Genomics Center for Infectious Disease (SSGCID) but was otherwise uncharacterized. ChMAT (PDB: 4ODJ) and hMAT2a (PDB: 5A1G) have 59% sequence identity and their asymmetric units are structurally conserved with a C-α alignment giving a RSMD of 0.47. A comparison between their overall fold and active sites is shown in Fig. S1 (ESI†). We assessed the MAT activity using methionine and ATP (2) – or ATP analogues 3–8 (Fig. 2) – by detecting the formation of AdoMet (9a) or AdoMet analogues (10a–15a), respectively, by LC-MS (Fig. S2–S8, ESI†). Next, we set up enzymatic cascade reactions with the highly promiscuous MTase Ecm118,32 to evaluate how efficiently the six different AdoMet analogues are formed, avoiding effects from the strong product inhibition of MAT (Fig. 3).
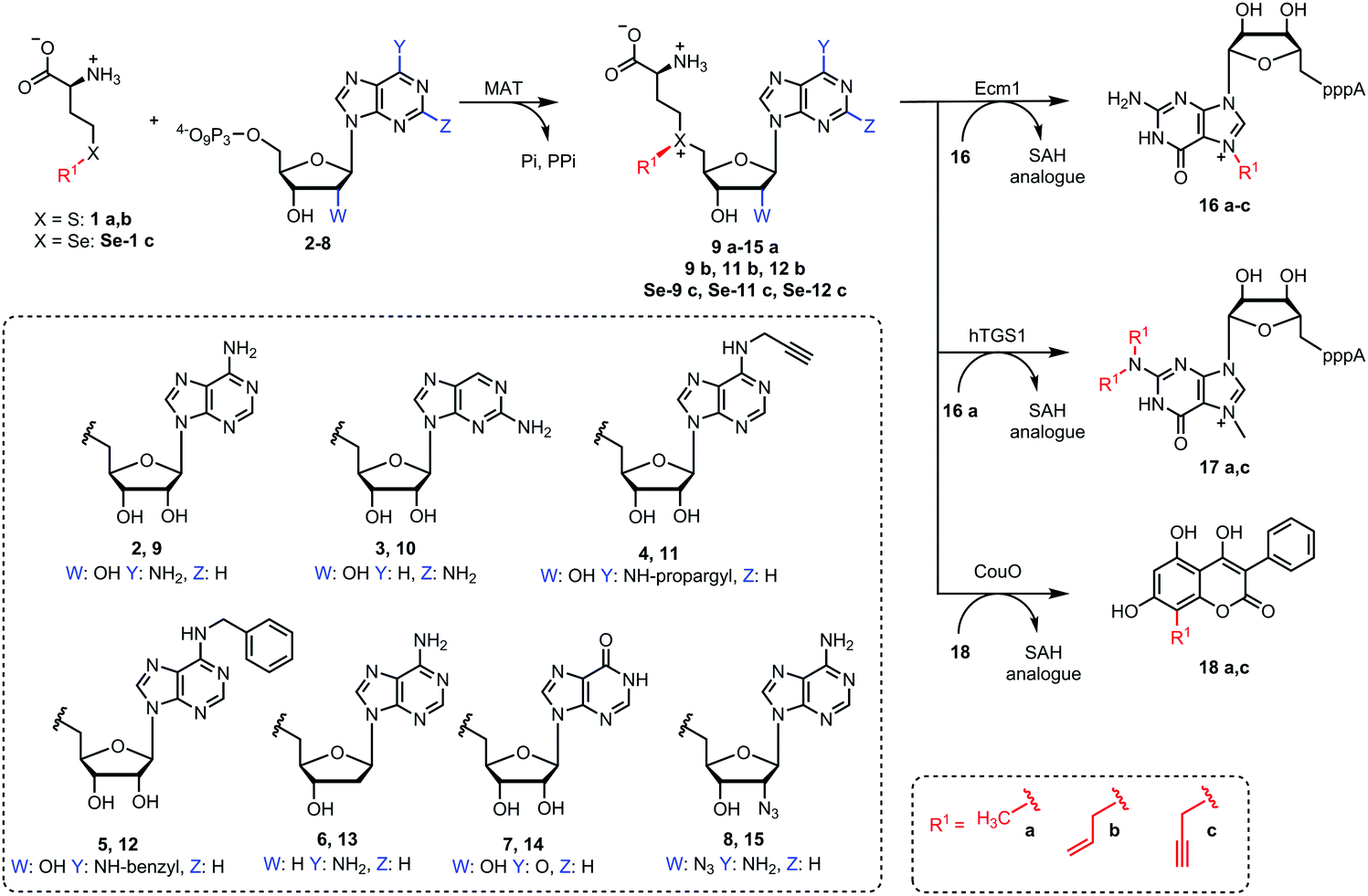 | ||
| Fig. 2 Methionine (1a) is enzymatically converted by MAT with ATP (2) or different nucleoside triphosphates (3–8) to form AdoMet (9a) or NM-AdoMet analogues (10–15a) with modifications in the nucleoside moiety. These NM-AdoMet analogues are accepted by methyltransferases as shown for Ecm1 in an enzymatic cascade reaction. Furthermore, methionine analogues (1b, Se-1c) and ATP analogues (4, 5) can be converted to AdoMet analogues (11b, Se-11c, 12b and Se-12c) by MAT enzymes. These AdoMet analogues provide selectivity among MTase catalyzed alkylation reactions. | ||
 | ||
| Fig. 3 Methionine (1a) can be reacted with ATP analogues (2–8) by ChMAT (A) or hMAT2a I117A (B). The resulting AdoMet analogues 9a–15a can be used as cofactors by a suitable methyltransferase as shown here for Ecm1. Conditions: 6 mM 1a, 2.5 mM 2–8, 1 mol% MAT (rel. to 2–8), 300 μM 16, 10 mol% Ecm1 (rel. to 16). The reaction mixture was incubated at 23 °C and analyzed by HPLC at time points indicated. | ||
With the canonical substrates methionine (1a) and ATP (2) generating 9a, GpppA (16) was rapidly and completely reacted using both MAT/Ecm1 combinations, yielding ≥99% 16a after 2 h reaction time (Fig. 3A, B and Fig. S9, ESI†), confirming that both MAT enzymes are active. In addition, several of the other NTPs we tested gave high conversion under the same conditions. In particular, AdoMet analogues 10a and 11a derived from 2-aminopurine triphosphate (3) and N6-propargyl-ATP (4) led to the formation of 16a in near quantitative yield when ChMAT was used (Fig. 3A). Similarly, AdoMet analogues 14a and 12a derived from inosine triphosphate (7) and N6-benzyl-ATP (5) yielded up to 90% and 80% of 16a, respectively. Moderate yields were obtained with ChMAT when 2′-deoxy-ATP (6) was used to form an AdoMet analogue 13a (Fig. 3). The 2′-deoxy-2′-azido-ATP (8a) producing 15a gave low yields with ChMAT.
For hMAT2-Var, good yields were also observed for 10a, 12a and 11a (Fig. 3B). However, 13a, 14a and 15a were barely tolerated, yielding only 8–10% of product. Control reactions did not lead to the formation of 16a (Fig. S10, ESI†). These data show that the nucleoside moiety of ATP can be altered and still be converted by the two enzymes required for AdoMet generation (MAT) and methyl transfer (MTase). The activities obtained with hMAT2a-Var and wildtype ChMAT as well as the co-substrate promiscuity differ, showing that the putative ChMAT is not only an active enzyme but also more tolerant than hMAT2a-Var regarding alterations in the nucleoside moiety.
Next, we asked whether these NM-AdoMets could provide a new and complementary strategy to obtain differential activities for different MTases. We assembled a representative test set for MTases, namely Ecm1, hTGS1 and CouO based on their crystal structures (Fig. 4E–G).40–42 We performed cascade reactions with ChMAT using (i) the native substrates (1a and 2) forming 9a, (ii) a combination with modification at the Se-center (Se-1c and 2) forming Se-9c, or (iii) a combination with modification at the N6 position (1a and 5) forming 12a as the cosubstrate for the MTases. HPLC-analysis was performed at 30 min and 8 h by HPLC to ensure evaluation in the linear range as well as at maximum conversions (Fig. S11–S13, ESI†). For the natural substrates 1a and 2 and in situ formation of 9a, all three cascade reactions gave near quantitative conversion after 8 h (Ecm1 100%, hTGS1 89% and CouO 100%) (Fig. 4B–D). For Se-9c as the cosubstrate, two out of the three MTases retained high activity. Specifically, CouO yielded complete (100%) and Ecm1 high (87%) conversion after 8 h. The MTase hTGS1 however yielded only 5% conversion when Se-9c was produced as the cosubstrate (Fig. 4C).
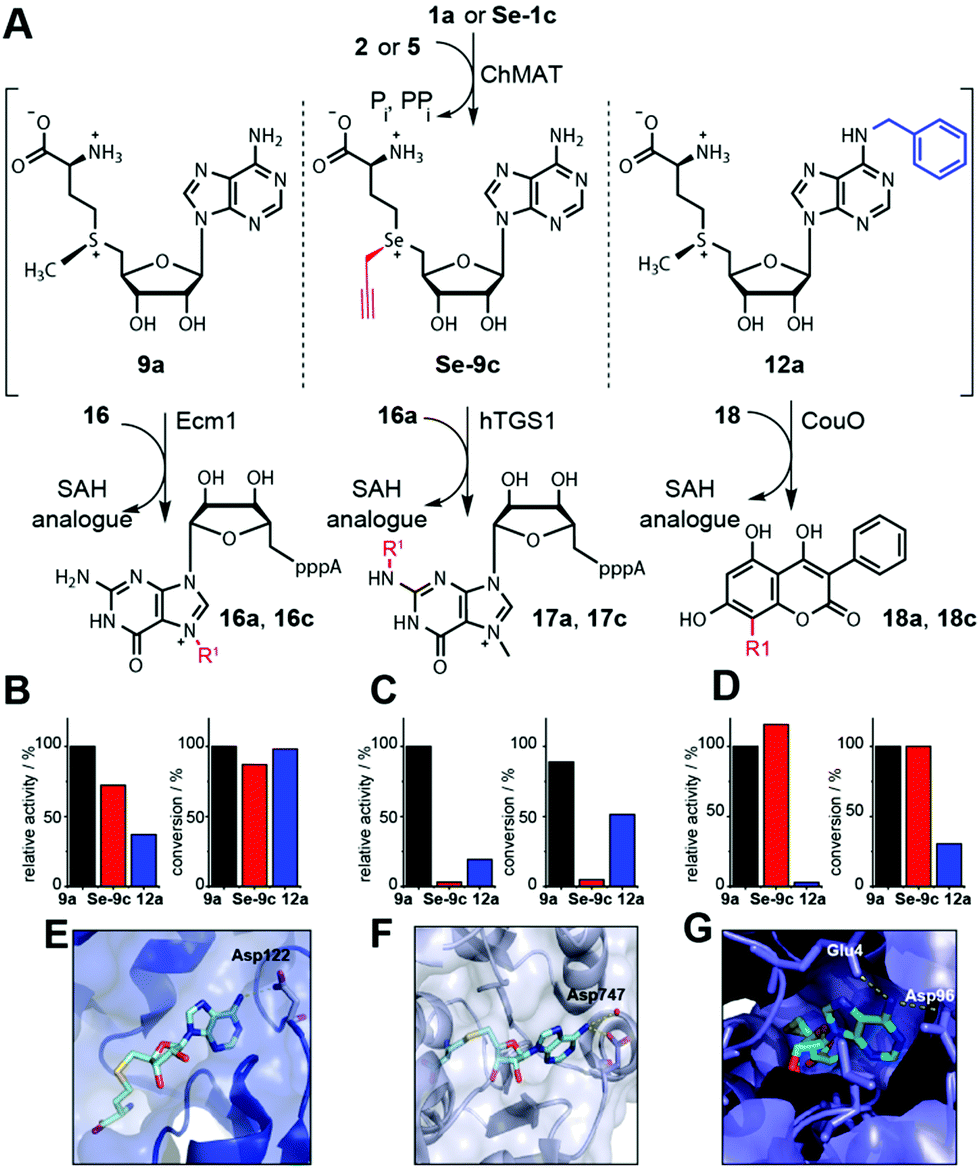 | ||
| Fig. 4 Differential MTase targeting by AdoMet analogues bearing no modification, a modification at the sulfur/selenium atom or at the nucleoside. (A) Reaction scheme. (B–D) Relative activities and maximum conversions for indicated AdoMet analogues for Ecm1 (B), hTGS1 (C) or CouO (D), respectively. Conditions: 6 mM 1a or Se-1c, 2.5 mM 2 or 5, 4 mol% ChMAT (rel. to 2 or 5), 300 μM 16, 16a, or 18, 10 mol% MTase (rel. to 16, 16a, or 18). The reaction mixture is incubated at 23 °C for 30 min (relative activities) or 8 h (conversions). (E–G) Crystal structures of Ecm1 (PDB: 1RI1), hTGS1 (PDB: 3GDH) and CouO (PDB: 5M58). For SAH (cyan sticks) polar interactions of the N6 position with the enzyme are highlighted in yellow dashed lines. In (E–G), the residues coordinating the N6 position of SAH are labeled. | ||
For NM-AdoMet 12a as the cosubstrate, a different activity profile was observed for the three MTases. Again, Ecm1 retained high conversion (98%), however, CouO showed only low conversion (30%). Here, hTGS1 – although almost inactive on Se-9c – was able to accommodate the N6-modification in 12a and showed 51% conversion after 8 h. Looking at the relative activities, it is evident that modifications at the S/Se atom (Se-9c) and modifications in the nucleoside (12a) are not equally well accommodated by different MTases. In line with the conversions, Se-9c drastically reduces the relative activity of hTGS1 to 3%, whereas CouO (115%) shows even higher relative activity and Ecm1 (72%) remains highly active. Thus, up to 37-fold discrimination was achieved for one of the three MTases in this test set. However, the activities of both Ecm1 and CouO remained high, limiting this type of AdoMet analogues for differential targeting. The N6-benzyl group reduced the relative activity of CouO to 3%, whereas the relative activities of Ecm1 (37%) and hTGS1 (19%) were less affected (Fig. 4B–D). Thus, the NM-AdoMet 12a leads to a nearly 13-fold difference in relative activities between Ecm1 and CouO allowing for discrimination where the S/Se-substituent had no effect.
Finally, we explored the possibility to produce and convert AdoMet analogues that carry modifications both at the S/Se atom and at the nucleoside moiety. We therefore tested whether ChMAT was also able to convert methionine analogues in combination with N6-modified ATPs. Specifically, we tested allyl-homocysteine (1b) and propargyl-selenohomocysteine (Se-1c) in addition to methionine (1a) in an enzymatic cascade with Ecm1 using 16 to obtain N7-alkylated products (16a–c) using N6-propargyl- (4) or N6-benzyl-ATP (5) as cosubstrates, instead of the natural ATP (Fig. S14, ESI†). Formation of the respective AdoMet-analogues (11b, Se-11c as well as 12b and Se-12c) was confirmed by LC-MS (Fig. S15–S18, ESI†).
Time courses showed that methionine analogues 1b and Se-1c were accepted by ChMAT—even when combined with N6-modified-ATPs. Cascade reactions with ChMAT/Ecm1 yielded up to 28% of N7-allyl-GpppA (16b) or 24% of N7-propargyl-GpppA (16c), respectively, when N6-propargyl ATP (4) was used (Fig. S14B, ESI†). With N6-benzyl ATP (5) the reaction yielded 20% N7-allyl-GpppA (16b) or 15% N7-propargyl-GpppA (16c), respectively (Fig. S14C, ESI†). When combining the enzymatic generation of Se-12c with different MTases, we found no activity in cascades with hTGS1 (0%) and low activity with CouO (4%) (Fig. S19, ESI†). These data show (i) that N6-modified ATPs can also be used in combination with methionine analogues to enzymatically generate the respective double modified AdoMets and (ii) that the latter are suitable co-substrates for site-specific transfer of different types of alkyl groups to an MTase target molecule.
Both MATs tolerate a number of ATP analogues and can be used in enzymatic cascades to generate the respective nucleoside-modified AdoMet analogues (NM-AdoMets) but the newly characterized ChMAT appears to be more promiscuous. Extending the size of the substituent at the sulfur/selenium atom can alter the relative activity of MTases and thus provide selectivity for a subset of MTases, however, this subset of MTases is rather large. Modifying the nucleoside moiety of AdoMet (12a) leads to selectivity for a different subset of MTases. The NM-AdoMet analogs represent a new strategy to address the MTase selectivity problem and – in combination with protein engineering in a “bump-and-hole” approach – exhibit potential to solve it.
A. R. thanks the ERC (772280) and the DFG (SFB858, IRTG 2027) for financial support. We thank A.-M. Lawrence-Dörner, A. Rahe, S. Wulff and Dr W. Dörner for excellent technical assistance. The MS and NMR facilities of the OC department are gratefully acknowledged for analytical services. The plasmid for ChMAT herein was provided by the SSGCID (http://www.SSGCID.org) which is supported by Federal Contract No. HHSN272201700059C from the NIAID, NIH and HHS.
Conflicts of interest
There are no conflicts to declare.References
- M. Fontecave, M. Atta and E. Mulliez, Trends Biochem. Sci., 2004, 29, 243–249 CrossRef CAS PubMed.
- F. R. Traube and T. Carell, RNA Biol., 2017, 14, 1099–1107 CrossRef PubMed.
- B. X. S. Zhao, I. A. Roundtree and C. He, Nat. Rev. Mol. Cell Biol., 2017, 18, 31–42 CrossRef CAS PubMed.
- G. Zheng, Y. Fu and C. He, Chem. Rev., 2014, 114, 4602–4620 CrossRef CAS PubMed.
- M. R. Bennett, S. A. Shepherd, V. A. Cronin and J. Micklefield, Curr. Opin. Chem. Biol., 2017, 37, 97–106 CrossRef CAS PubMed.
- C. Dalhoff, G. Lukinavicius, S. Klimasauakas and E. Weinhold, Nat. Protoc., 2006, 1, 1879–1886 CrossRef CAS PubMed.
- C. Dalhoff, G. Lukinavicius, S. Klimasauskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS PubMed.
- G. Lukinavičius, V. Lapienė, Z. Staševskij, C. Dalhoff, E. Weinhold and S. Klimašauskas, J. Am. Chem. Soc., 2007, 129, 2758–2759 CrossRef PubMed.
- W. Peters, S. Willnow, M. Duisken, H. Kleine, T. Macherey, K. E. Duncan, D. W. Litchfield, B. Luscher and E. Weinhold, Angew. Chem., Int. Ed., 2010, 49, 5170–5173 CrossRef CAS PubMed.
- M. Tomkuviene, B. Clouet-d'Orval, I. Cerniauskas, E. Weinhold and S. Klimasauskas, Nucleic Acids Res., 2012, 40, 6765–6773 CrossRef CAS PubMed.
- A. Osipenko, A. Plotnikova, M. Nainyte, V. Masevicius, S. Klimasauskas and G. Vilkaitis, Angew. Chem., Int. Ed., 2017, 56, 6507–6510 CrossRef CAS PubMed.
- C. Sommer-Kamann, A. Fries, S. Mordhorst, J. N. Andexer and M. Muller, Angew. Chem., Int. Ed., 2017, 56, 4033–4036 CrossRef CAS PubMed.
- M. Heimes, L. Kolmar and C. Brieke, Chem. Commun., 2018, 54, 12718–12721 RSC.
- J. Deen, C. Vranken, V. Leen, R. K. Neely, K. P. F. Janssen and J. Hofkens, Angew. Chem., Int. Ed., 2017, 56, 5182–5200 CrossRef CAS PubMed.
- J. M. Holstein, D. Stummer and A. Rentmeister, Chem. Sci., 2015, 6, 1362–1369 RSC.
- J. M. Holstein, D. Schulz and A. Rentmeister, Chem. Commun., 2014, 50, 4478–4481 RSC.
- D. Schulz, J. M. Holstein and A. Rentmeister, Angew. Chem., 2013, 52, 7874–7878 CrossRef CAS PubMed.
- F. Muttach, F. Mäsing, A. Studer and A. Rentmeister, Chem. – Eur. J., 2017, 23, 5988–5993 CrossRef CAS PubMed.
- R. Wang, W. Zheng, H. Yu, H. Deng and M. Luo, J. Am. Chem. Soc., 2011, 133, 7648–7651 CrossRef CAS PubMed.
- S. Kim, A. Gottfried, R. R. Lin, T. Dertinger, A. S. Kim, S. Chung, R. A. Colyer, E. Weinhold, S. Weiss and Y. Ebenstein, Angew. Chem., Int. Ed., 2012, 51, 3578–3581 CrossRef CAS PubMed.
- L. Anhauser, F. Muttach and A. Rentmeister, Chem. Commun., 2017, 54, 415–572 Search PubMed.
- J. M. Holstein, L. Anhäuser and A. Rentmeister, Angew. Chem., 2016, 55, 10899–10903 CrossRef CAS PubMed.
- F. Muttach and A. Rentmeister, Methods, 2016, 107, 3–9 CrossRef CAS PubMed.
- F. Muttach and A. Rentmeister, Angew. Chem., 2016, 55, 1917–1920 CrossRef CAS PubMed.
- S. Singh, J. J. Zhang, T. D. Huber, M. Sunkara, K. Hurley, R. D. Goff, G. J. Wang, W. Zhang, C. M. Liu, J. Rohr, S. G. Van Lanen, A. J. Morris and J. S. Thorson, Angew. Chem., Int. Ed., 2014, 53, 3965–3969 CrossRef CAS PubMed.
- M. Dippe, W. Brandt, H. Rost, A. Porzel, J. Schmidt and L. A. Wessjohann, Chem. Commun., 2015, 51, 3637–3640 RSC.
- M. Zhao, Y. S. Wijayasinghe, P. Bhansali, R. E. Viola and R. M. Blumenthal, Microbiology, 2015, 161, 674–682 CrossRef CAS PubMed.
- R. Wang, K. Islam, Y. Liu, W. Zheng, H. Tang, N. Lailler, G. Blum, H. Deng and M. Luo, J. Am. Chem. Soc., 2013, 135, 1048–1056 CrossRef CAS PubMed.
- K. Hartstock, B. S. Nilges, A. Ovcharenko, N. V. Cornelissen, N. Püllen, A. M. Lawrence-Dörner, S. A. Leidel and A. Rentmeister, Angew. Chem., 2018, 57, 6342–6346 CrossRef CAS PubMed.
- R. Wang, W. Zheng and M. Luo, Anal. Biochem., 2014, 450, 11–19 CrossRef CAS PubMed.
- The UniProt Consortium, Nucleic Acids Res., 2017, 45, D158–D169 CrossRef PubMed.
- F. Muttach, N. Muthmann, D. Reichert, L. Anhauser and A. Rentmeister, Chem. Sci., 2017, 8, 7947–7953 RSC.
- G. Lukinavičius, M. Tomkuvienė, V. Masevičius and S. Klimašauskas, ACS Chem. Biol., 2013, 8, 1134–1139 CrossRef PubMed.
- Y. Liu, K. Shah, F. Yang, L. Witucki and K. M. Shokat, Chem. Biol., 1998, 5, 91–101 CrossRef CAS PubMed.
- M. A. Shogren-Knaak, P. J. Alaimo and K. M. Shokat, Annu. Rev. Cell Dev. Biol., 2001, 17, 405–433 CrossRef CAS PubMed.
- M. Ellermann, R. Jakob-Roetne, C. Lerner, E. Borroni, D. Schlatter, D. Roth, A. Ehler, M. G. Rudolph and F. Diederich, Angew. Chem., Int. Ed., 2009, 48, 9092–9096 CrossRef CAS PubMed.
- Q. Lin, F. Y. Jiang, P. G. Schultz and N. S. Gray, J. Am. Chem. Soc., 2001, 123, 11608–11613 CrossRef CAS PubMed.
- I. McKean, J. Sadler, A. Cuetos, A. Frese, L. Humphreys, G. Grogan, P. Hoskisson and G. Burley, Angew. Chem., Int. Ed., 2019, 58, 17583–17588 CrossRef CAS PubMed.
- R. Wang, K. Islam, Y. Liu, W. Zheng, H. Tang, N. Lailler, G. Blum, H. Deng and M. Luo, J. Am. Chem. Soc., 2013, 135, 1048–1056 CrossRef CAS PubMed.
- S. Hausmann, S. S. Zheng, C. Fabrega, S. W. Schneller, C. D. Lima and S. Shuman, J. Biol. Chem., 2005, 280, 20404–20412 CrossRef CAS PubMed.
- T. Monecke, A. Dickmanns and R. Ficner, Nucleic Acids Res., 2009, 37, 3865–3877 CrossRef CAS PubMed.
- H. Stecher, M. Tengg, B. J. Ueberbacher, P. Remler, H. Schwab, H. Griengl and M. Gruber-Khadjawi, Angew. Chem., Int. Ed., 2009, 48, 9546–9548 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc07807j |
| This journal is © The Royal Society of Chemistry 2020 |