
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater envelope has a critical impact on the design of protein–protein interaction inhibitors†
Ekaterina L.
Ratkova
 *ab,
Maciej
Dawidowski
ac,
Valeria
Napolitano
d,
Grzegorz
Dubin
d,
Roberto
Fino
a,
Michael S.
Ostertag
a,
Michael
Sattler
ae,
Grzegorz
Popowicz
*a and
Igor V.
Tetko
*af
*ab,
Maciej
Dawidowski
ac,
Valeria
Napolitano
d,
Grzegorz
Dubin
d,
Roberto
Fino
a,
Michael S.
Ostertag
a,
Michael
Sattler
ae,
Grzegorz
Popowicz
*a and
Igor V.
Tetko
*af
aInstitute of Structural Biology, Helmholtz Zentrum München – German Research Center for Environmental Health (GmbH), Ingolstädter Landstraße 1, 85764 Neuherberg, Germany. E-mail: grzegorz.popowicz@helmholtz-muenchen.de; i.tetko@helmholtz-muenchen.de
bMedicinal Chemistry, Cardiovascular, Renal and Metabolic Diseases, IMED Biotech Unit, AstraZeneca, Gothenburg, Sweden. E-mail: ekaterina.ratkova@astrazeneca.com
cDepartment of Drug Technology and Pharmaceutical Biotechnology, Medical University of Warsaw, Banacha 1, 02-097 Warszawa, Poland
dFaculty of Biochemistry, Biophysics and Biotechnology and Malopolska Centre of Biotechnology, Jagiellonian University, Gronostajowa 7, 30-387 Krakow, Poland
eCenter for Integrated Protein Science Munich at Chair of Biomolecular NMR, Department Chemie, Technische Universität München, Lichtenbergstrasse 4, 85747 Garching, Germany
fBIGCHEM GmbH, Valerystr. 49, 85716 Unterschleißheim, Germany
First published on 20th March 2020
Abstract
We show that a water envelope network plays a critical role in protein–protein interactions (PPI). The potency of a PPI inhibitor is modulated by orders of magnitude on manipulation of the solvent envelope alone. The structure–activity relationship of PEX14 inhibitors was analyzed as an example using in silico and X-ray data.
Over the last decade, the analysis of water molecules solvating protein–ligand binding surfaces was demonstrated to be highly valuable for drug design.1,2 The most widely employed application involves high-energy ‘unhappy’ water molecules, which are situated in deep lipophilic pockets and are often isolated from the rest of the solvation shell. Their number, arrangement and proximity were found to be closely connected to the druggability of the corresponding receptor part.3 Commonly, drug candidates, which were designed to displace ‘unhappy’ water molecules significantly benefit from binding enthalpy.4–6 Another case involves energetically favourable ‘happy’ water molecules. These tightly interact with polar surface residues and are often involved in a network with other water molecules in the solvation shell. During a binding event, ‘happy’ water molecules can support protein–ligand complementarity via mediating their interactions.7,8 Such water-bridges have always been considered as an important feature in drug design: a ligand-based project could be complicated due to a poor overlap between ligand pharmacophore models, while a structure-based project could suffer because of the lack of reproducibility of X-ray data.9 Despite numerous studies, the utility of a displacement of ‘happy’ water molecules is still a matter of discussion.10,11
In case of protein–protein interactions (PPIs), the impact of water molecules could be even more sophisticated. Due to a large shallow and solvent-exposed binding surface, PPIs are often considered as a unique challenge for drug design.12 Recently, Cramer and co-workers reported that inhibition of PPIs with small molecules could be enhanced by optimization of the water network wrapping a newly formed complex surface, ‘water envelope’.13 A better adaption of water molecules to an energetically favourable architecture of the interaction network, the higher is the potency of the ligand. For example, a ≈50-fold increase in affinity was reported for thermolysin inhibitors.14
As a proof-of-concept study, we studied the inhibition of the PPI involving the T.brucei PEX14 protein and the α-helical motif of PEX5, which impairs the trypanosome viability.15 We showed that the direct binding of small-molecule ligands (Fig. 1, right) to PEX14 is capable of disrupting its interaction with PEX5, and can efficiently kill trypanosome parasites.15 Structural data indicated an intricate character of the system where water molecules could have a critical impact on the interactions. There are no directional interactions between the ligand and the receptor: binding is driven by non-polar interactions, while all contacts with polar groups are water-mediated (Fig. 1, left top). Apart from that, interactions with “hot spot” cavities were not limited by the lipophilicity-driven structure–activity relationship (SAR) (Fig. 1, left bottom).
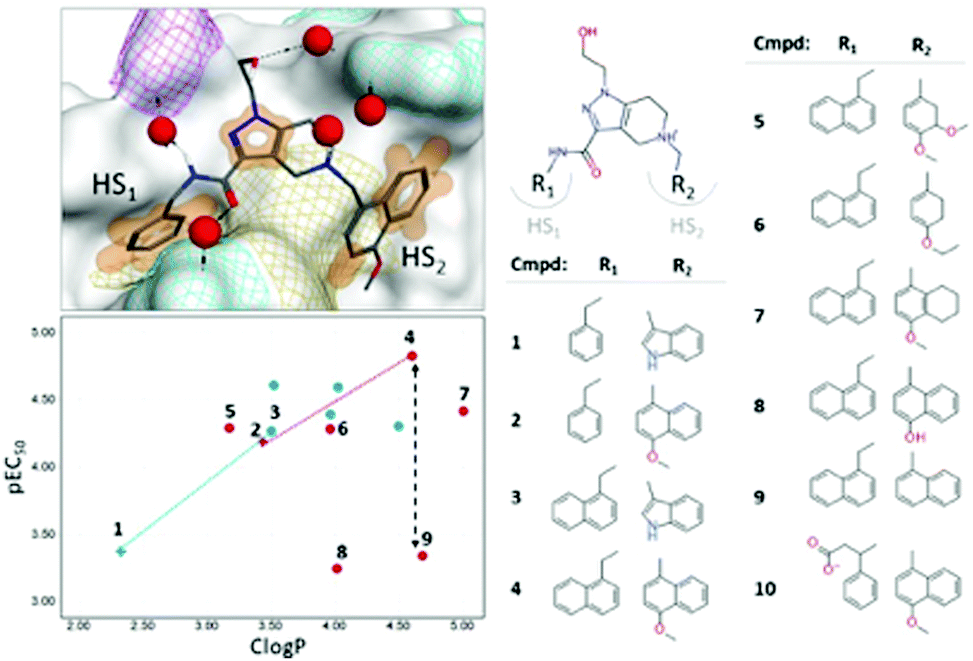 | ||
| Fig. 1 Right: Selected set of PEX14 inhibitors (see also ref. 16). Left top: Structure of the complex of the T.brucei PEX14 protein and compound 2 (PDB ID = 5L8A). The protein is represented with a van der Waals surface colored by yellow – lipophilic, cyan – positively charged, magenta – negatively charged. The orange surface shows non-polar interactions. Water molecules near the binding surface are indicated with red spheres. Left bottom: SAR on R1- and R2-groups of the inhibitors, data points are shaped with respect to R1-group (empty cross – phenyl group, filled circle – naphthyl group) and colored by type of R2-group (blue – indole ring, red – benzyl/naphthyl ring). Dotted lines indicate lipophilicity-driven binding in cavity HS1, while black dashed arrow showed spread in potency for iso-lipophilic compounds in HS2 pocket. | ||
To investigate the solvation effects in the PEX14 – inhibitor complex and check if a water envelope plays a significant role in the inhibition, we computationally assessed the interaction surface. The following features were analyzed: (i) geometrical: radial distribution functions (RDFs, i.e. the probability to find a water molecule at a particular distance from a solute) and explicit water positions defined from RDFs and (ii) energetical: free energy maps (free energy change corresponding to a water transfer from a particular position around the solute to a bulk solvent). All parameters were calculated with a 3D Reference Interacting Site Model (3D-RISM) implemented in MOE software (Chemical Computing Group).17 In a number of publications it was shown that this physically rigorous computational approach reproduced with high accuracy experimental data on the structure of solvation shells for various systems.18–20 For PEX14 structures, we revealed an excellent agreement between the predicted water positions and high-resolution X-ray data (PDB ID = 5L87; res. = 0.87 Å, see Fig. S8, ESI†). This observation gave us a solid basis to use the same approach for the analysis of the ligand-free receptor, where crystal structures could not be obtained. Details of the 3D-RISM calculation setup and data analysis are provided in the ESI.† For compounds lacking X-ray structures in the retrospective study as well as for systems in the prospective study, we performed docking using the template-docking protocol implemented in MOE software (details of the protocol setup are provided in the ESI†).
In case of PPIs, it is common that interactions within “hot-spot” (HS) cavities contribute strongest to the binding. Therefore, modifications of ligand's groups involved in the interactions lead to largest changes in the affinity.21 We observed that binding to cavity HS1 is purely lipophilicity-driven (the chemical composition of the R1-group is shown in Fig. S9, ESI†). The most pronounced change in affinity, by one log-unit, is associated with the growth of the phenyl ring to the naphthyl one: molecular pairs 1–3, and 2–4 (Fig. 1, bottom left, empty crosses). In contrast, the SAR on the R2-group approaching the HS2 cavity is rather flat with non-obvious outliers (Fig. 1, bottom left, filled circles).21 We considered two types of R2-groups, based on (i) indole and (ii) benzyl/naphthyl rings. In both cases, an increase in lipophilicity does not influence the potency within a series (whole indole series and compounds 5, 6, and 7 from benzyl/naphthyl series). However, specific structural changes lead to iso-lipophilic compounds with quite variable affinity values (benzyl/naphthyl series: molecular pairs 6–8 and 4–9). The solvent analysis of the HS2 cavity revealed that water patterns are different for indole and naphthyl rings (Fig. S10, ESI†). Upon binding of the indole ring, several water molecules remained bound to the cavity, whereas the methoxynaphthyl moiety efficiently displaced all water molecules from the HS2 cavity. This observation agrees with the routine practice of handing unfavorable water molecules. Notably, the binding of the methoxynaphthyl fragment was accompanied by a change in the conformation of the Thr22 residue, which allowed an optimal shape match between the cavity and R2-group (Fig. S10, ESI†). It is in line with a lipophilicity-driven change in binding for molecular pairs 1–2 and 3–4 (Fig. 1, bottom left). For extreme cases of reduced-potency compounds (8 and 9), we revealed that very energetically unfavorable water molecules remained in the cavity after binding (Fig. S10, ESI†).
The solvent analysis in hot-spot cavities is summarized as follows: (i) binding of inhibitors to the buried and narrow HS1 cavity was purely lipophilicity-driven, while their binding to shallow HS2 cavity had a more complicated character, (ii) in the absence of a ligand, the HS2 cavity could accommodate a network of 3–4 water molecules forming H-bonds with Ser26, Thr22, and Agr18 residues, and (iii) if any of the water molecules remained in the cavity after a binding event, their energetic profile heavily influences the inhibition. The most pronounced gain in potency was archived by compound 4, which has the best shape complementarity with the receptor leading to both, the most efficient non-polar interactions and displacement of all water molecules from the cavity.
To our surprise, an even more impressive SAR with a comparable boost in potency was observed for a water-exposed surface of the receptor (situated between HS1 and HS2 cavities). We considered that a solvent analysis of this part of the receptor could help in better understanding of the water envelope and its role in inhibition. Here, all contacts between the ligand and receptor are mediated by 1–2 water molecules (Fig. 1, left top). Solvent analysis of free and bound states of both the receptor and ligand revealed that the water molecules belonged mainly to receptor's solvation shell. In a complex, they remained tightly coordinated by polar residues (Fig. 2, middle) and are required for the adaptation of the ligand to a large flat receptor surface with rather remote polar regions (Fig. 2, right). Attempts to disrupt the water-bridges yielded significantly less active inhibitors (Fig. 3). When a water molecule has the same position in solvation shells of both the ligand and receptor in their binding conformation, the corresponding water bridge is particularly energetically favorable.
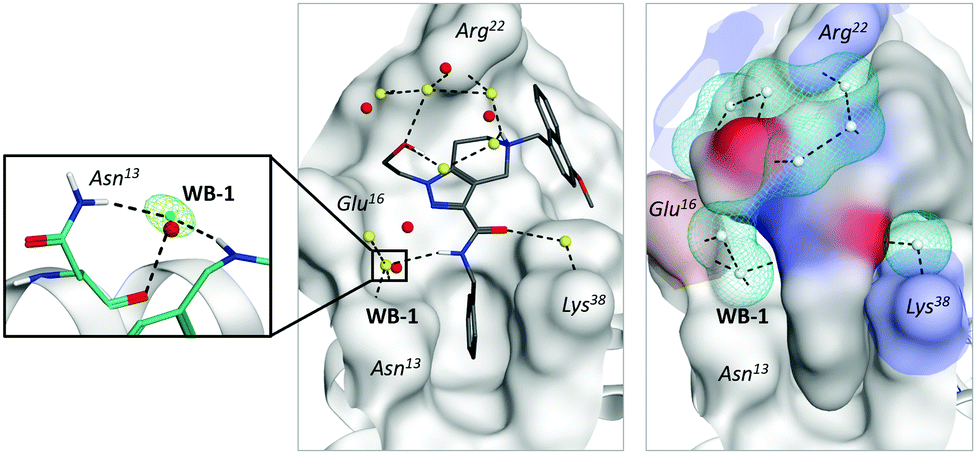 | ||
| Fig. 2 Water-mediated interactions between PEX14 and ligand 2 (PDB ID = 5L8A). Middle: X-ray water molecules are shown in red, while predicted water molecules are shown in yellow. Right: Shape complementarity between the ligand and receptor (color patches represented polar residues), cyan meshed surfaces indicated predicted water positions). Left: Coordination of the most energetically stable water bridge (WB-1). | ||
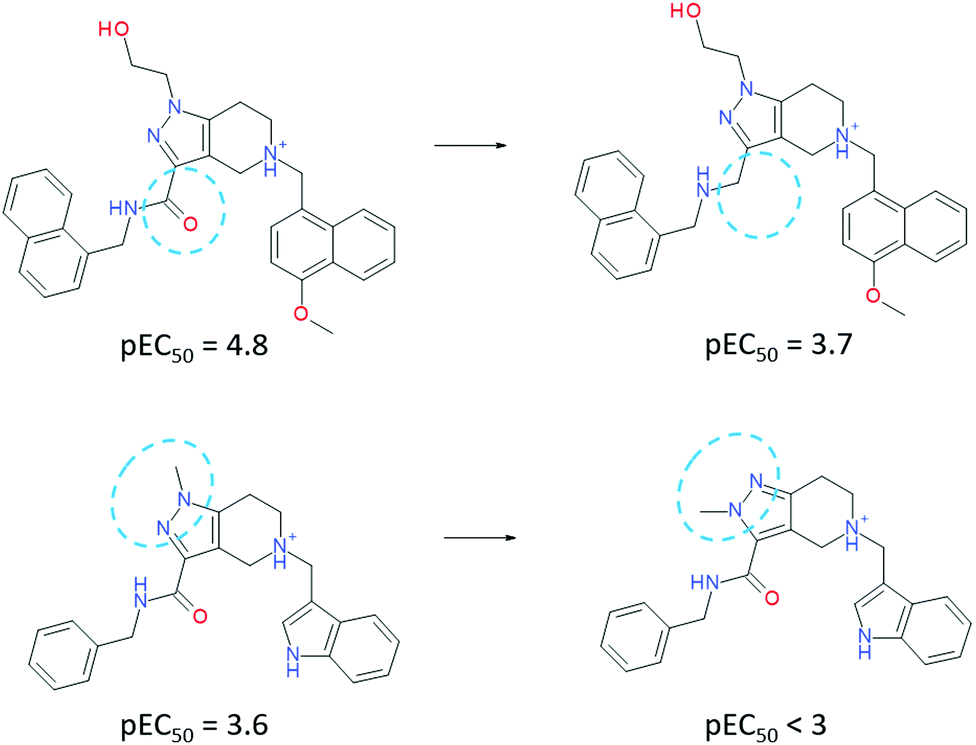 | ||
| Fig. 3 Matched molecular pairs of ligands, which exhibit high importance of the water-mediated interactions. | ||
We observed that one water molecule was highly conserved in all X-ray structures of the inhibitor series (Fig. 2, left).7 It mediated the interactions between the amide-group of the ligand and peripheral Asn31 residue of the protein. The particularly conserved position of this water molecule is also related to the coordination of the backbone and side-chain of the same residue.
According to our model, water molecules mediating protein–ligand interactions in different parts of the binding surface were further interconnected between each other by energetically favourable water molecules forming an extended network – water envelope (Fig. 4, left, black meshed surface). To evaluate its impact, we suggest the following structural modifications. Two spatially separated parts of the network (around Asn13 and Lys38 residues) could be connected via a carboxylate group (compound 10). The introduction of the group yielded two enantiomers, where the (S)-isomer gained in potency and the (R)-isomer only weakly interacted with PEX14: parent compound 2 pIC50 = 4.2, (S)-isomer pIC50 = 4.8, and (R)-isomer pIC50 = 3.2. Notably, experimental X-ray structures of the (S)-isomer (PDB ID = 5OML) showed that it formed two additional water-bridges with the receptor (Fig. 4, middle, WB-2 and WB-3), which allowed more interconnections within the water envelope. We also determined the X-ray structure of the weakly inhibiting (R)-isomer (PDB ID = 6RT2). In this case, we observed that the carboxylic group has several possible positions including the one with the direct bond to Lys38 (Fig. 4, right). The direct interaction of the carboxyl group and primary amine was, however, not sufficient to compensate for the loss of optimal water network configuration. These results strongly support the hypothesis of Cramer and co-workers that the configuration of a water envelope could significantly affect the ligand potency.14
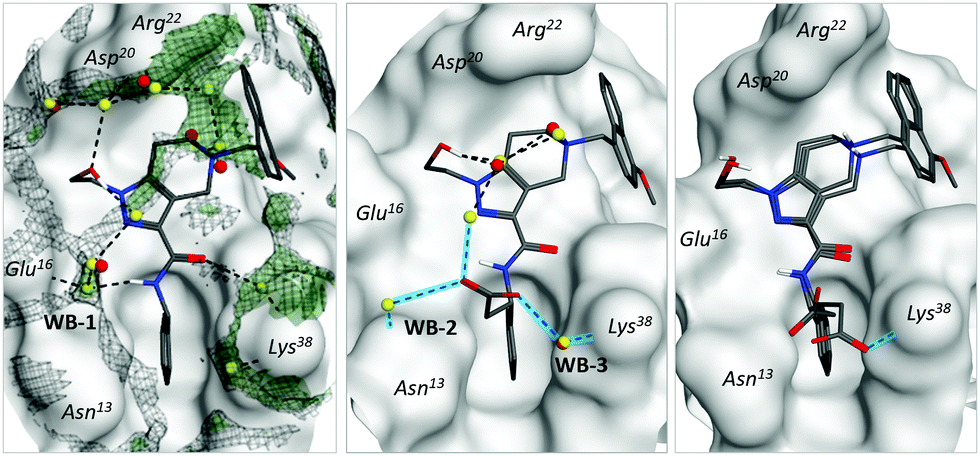 | ||
| Fig. 4 Water-mediated interactions between inhibitors and polar receptor residues. Crystallographic water molecules are shown in red, while predicted water molecules are shown in yellow. Left: Complex of PEX14 with ligand 2 (PDB ID = 5L8A). Meshed black surface corresponds to the radial distribution function g(r) > 3, green surface reflects the positions of energetically favorable water molecules (ΔGdes < −5 kcal mol−1). Middle: Complex of the protein with the S-isomer of ligand 10 (PDB ID = 5OML). Blue lines show newly formed bonds with respect to parent compound 2 (unaffected water bridges were skipped for simplicity). Right: Complex of the protein with the R-isomer of ligand 10 (PDB ID = 6RT2). Both possible positions of the carboxylic group are depicted. Notation is the same as the middle subfigure. | ||
We observed that the presence of water molecules at a large shallow PEX14 binding surface significantly modulates inhibitors’ activity leading to a flat unpredictable SAR. Remarkably, an optimally configured water envelope wrapping/mediating a primarily lipophilic complex is critical for binding energetics. Not only water molecules that are bridging the receptor with the ligand are important but also equally those water molecules that surround the interface. These molecules have little direct interaction with their binding partners, yet their optimal placement can yield significant improvement in binding. Therefore, studies on inhibition of PPIs’ interfaces should equally consider: (i) non-polar interactions within “hot-spot” cavities, (ii) water-bridges with polar surface residues, and (iii) a water envelope wrapping the newly formed complex. We suggest here a solvent analysis protocol based on 3D-RISM calculations as an efficient tool for the investigation of water envelopes in structure-based drug design.
ELR thanks the Alexander von Humboldt Foundation (Carl Friedrich von Siemens research award) for the financial support. MD acknowledges financial support from UMO-2018/31/B/NZ7/02089 and GD from UMO-2017/26/M/NZ1/00797, both from the National Science Center, Poland. This study received funding from the European Union's Framework Programme for Research and Innovation Horizon 2020 (2014–2020) under the Marie Skłodowska-Curie Grant Agreement No. 675555, Accelerated Early staGe drug discovery (AEGIS).
Conflicts of interest
Authors declare no conflict of interest.Notes and references
- P. Ball, Chem. Rev., 2008, 108, 74–108 CrossRef CAS PubMed.
- Schrodinger, http://www.schrodinger.com/science-articles/water-thermodynamics.
- J. S. Mason, A. Bortolato, M. Congreve and F. H. Marshall, Trends Pharmacol. Sci., 2012, 33, 249–260 CrossRef CAS PubMed.
- R. Abel, T. Young, R. Farid, B. J. Berne and R. A. Friesner, J. Am. Chem. Soc., 2008, 130, 2817–2831 CrossRef CAS PubMed.
- T. Beuming, Y. Che, R. Abel, B. Kim, V. Shanmugasundaram and W. Sherman, Proteins, 2012, 80, 871–883 CrossRef CAS PubMed.
- T. Beuming, R. Farid and W. Sherman, Protein Sci., 2009, 18, 1609–1619 CrossRef CAS PubMed.
- M. H. Ahmed, F. Spyrakis, P. Cozzini, P. K. Tripathi, A. Mozzarelli, J. N. Scarsdale, M. A. Safo and G. E. Kellogg, PLoS One, 2011, 6, e24712 CrossRef CAS PubMed.
- A. Kumar and K. Y. Zhang, J. Chem. Inf. Model., 2013, 53, 1880–1892 CrossRef CAS PubMed.
- D. R. Weiss, A. Bortolato, B. Tehan and J. S. Mason, J. Chem. Inf. Model., 2016, 56, 642–651 CrossRef CAS PubMed.
- R. Abel, N. K. Salam, J. Shelley, R. Farid, R. A. Friesner and W. Sherman, ChemMedChem, 2011, 6, 1049–1066 CrossRef CAS PubMed.
- R. Horbert, B. Pinchuk, E. Johannes, J. Schlosser, D. Schmidt, D. Cappel, F. Totzke, C. Schachtele and C. Peifer, J. Med. Chem., 2015, 58, 170–182 CrossRef CAS PubMed.
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Nat. Rev. Drug Discovery, 2016, 15, 533–550 CrossRef CAS PubMed.
- M. Betz, T. Wulsdorf, S. G. Krimmer and G. Klebe, J. Chem. Inf. Model., 2016, 56, 223–233 CrossRef CAS PubMed.
- J. Cramer, S. G. Krimmer, A. Heine and G. Klebe, J. Med. Chem., 2017, 60, 5791–5799 CrossRef CAS PubMed.
- M. Dawidowski, L. Emmanouilidis, V. C. Kalel, K. Tripsianes, K. Schorpp, K. Hadian, M. Kaiser, P. Maser, M. Kolonko, S. Tanghe, A. Rodriguez, W. Schliebs, R. Erdmann, M. Sattler and G. M. Popowicz, Science, 2017, 355, 1416–1420 CrossRef CAS PubMed.
- M. Dawidowski, V. C. Kalel, V. Napolitano, R. Fino, K. Schorpp, L. Emmanouilidis, D. Lenhart, M. Ostertag, M. Kaiser, M. Kolonko, B. Tippler, W. Schliebs, G. Dubin, P. Maser, I. V. Tetko, K. Hadian, O. Plettenburg, R. Erdmann, M. Sattler and G. M. Popowicz, J. Med. Chem., 2020, 63, 847–879 CrossRef CAS PubMed.
- MOE, Molecular Operating Environment (MOE), http://www.chemcomp.com/Products.htm.
- L. Fusani, I. Wall, D. Palmer and A. Cortes, Bioinformatics, 2018, 34, 1947–1948 CrossRef CAS PubMed.
- E. L. Ratkova, D. S. Palmer and M. V. Fedorov, Chem. Rev., 2015, 115, 6312–6356 CrossRef CAS PubMed.
- A. Kovalenko, Condens. Matter Phys., 2015, 18, 32601 CrossRef.
- T. Clackson and J. A. Wells, Science, 1995, 267, 383–386 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details and experimental procedure. See DOI: 10.1039/c9cc07714f |
| This journal is © The Royal Society of Chemistry 2020 |