
Au(I)/Au(III)-Catalyzed C–N coupling†
Jessica
Rodriguez
 a,
Nicolas
Adet
a,
Nathalie
Saffon-Merceron
b and
Didier
Bourissou
*a
a,
Nicolas
Adet
a,
Nathalie
Saffon-Merceron
b and
Didier
Bourissou
*a
aCNRS/Université Paul Sabatier, Laboratoire Hétérochimie Fondamentale et Appliquée (LHFA, UMR 5069), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France. E-mail: dbouriss@chimie.ups-tlse.fr
bInstitut de Chimie de Toulouse (FR 2599), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France
First published on 21st November 2019
Abstract
Cycling between Au(I) and Au(III) is challenging, so gold-catalyzed cross-couplings are rare. The (MeDalphos)AuCl complex, which we showed was prone to undergo oxidative addition, is reported here to efficiently catalyze the C–N coupling of aryl iodides and amines. The transformation does not require an external oxidant or a directing group. It is robust and works with a wide scope of aryl iodides and N-nucleophiles under mild conditions. Mechanistic studies, including the NMR and MS characterization of a key aryl amido Au(III) complex, strongly support a 2e redox cycle in which oxidative addition precedes transmetalation and reductive elimination is the rate-determining step.
Gold(III) chemistry has grown spectacularly during the last decade,1 but applications in catalysis are still rare, mainly due to the difficulty to access the +III oxidation state. Nevertheless, some C–C and C–X (X = N, O…) cross-coupling reactions proceeding via Au(I)/Au(III) catalytic cycles have been reported recently, in particular arylation reactions.2,3 Most of these transformations make use of a strong external oxidant or a diazonium salt as an electrophile, which limits the scope of compatible coupling partners and functional groups.4 Our group is exploring a complementary strategy based on “ancillary” ligands that emulate otherwise disfavoured reactivity at gold, such as oxidative addition.5,6 In this respect, the (P,N) ligand MeDalphos [Ad2P(o-C6H4)NMe2] was recently shown to readily promote oxidative addition of aryl iodides to gold and to enable catalytic C–C cross-coupling reactions under mild conditions without the need for an external oxidant.7,8 The (MeDalphos)AuCl complex was later reported by Spokoyny and coworkers to be very efficient for the stoichiometric arylation (C–S coupling) of unprotected peptides and proteins, as well as for the construction of hybrid nanoclusters.9
C–N cross-coupling is undoubtedly the most straightforward and general route to prepare aryl-amines. Palladium (Buchwald–Hartwig amination)10 and copper (Ullmann and Chan–Lam couplings)11 occupy forefront positions in transition metal-catalyzed C–N cross-coupling. Gold-catalyzed methodologies are very interesting to develop as the specific properties of gold may enable transformations to be achieved which are challenging or limited in scope with the other transition metals, but gold has been very rarely reported to promote C–N bond formation and C–N reductive elimination from Au(III) complexes actually remains very scarce.
As summarized in Fig. 1, a few arylation reactions of N-nucleophiles mediated by gold(III) have been reported over the last decade.12–16 Limbach and coworkers provided in 2010 the first examples of stoichiometric C–N reductive elimination of aryl gold(III) complexes by reacting well-defined 2,6-lutidine aryl gold(III) complexes with N-nucleophiles (Fig. 1a).12 Very recently, Toste et al. described C–N coupling between an aryldiazonium salt and succinimide mediated by gold under photoredox conditions and detailed mechanistic studies were performed (Fig. 1b).13 To the best of our knowledge, gold-catalyzed C–N couplings have only been reported twice so far. Direct and regioselective arylation of phthalimide (oxidative C–N coupling with arenes) has been achieved in 2015 by DeBoef and coworkers using phosphine gold(I) complexes and PhI(OAc)2 as an external oxidant.14 More recently, in 2017, Ribas et al. described the first oxidant-free Au-catalyzed C–N coupling involving aryl iodides (Fig. 1d).15 Despite these significant advances, the reaction conditions (temperature ≥100 °C, catalytic loading = 10 mol%) and the requirement of an external oxidant or a preinstalled directing group limit the scope and practical interest of the transformation.
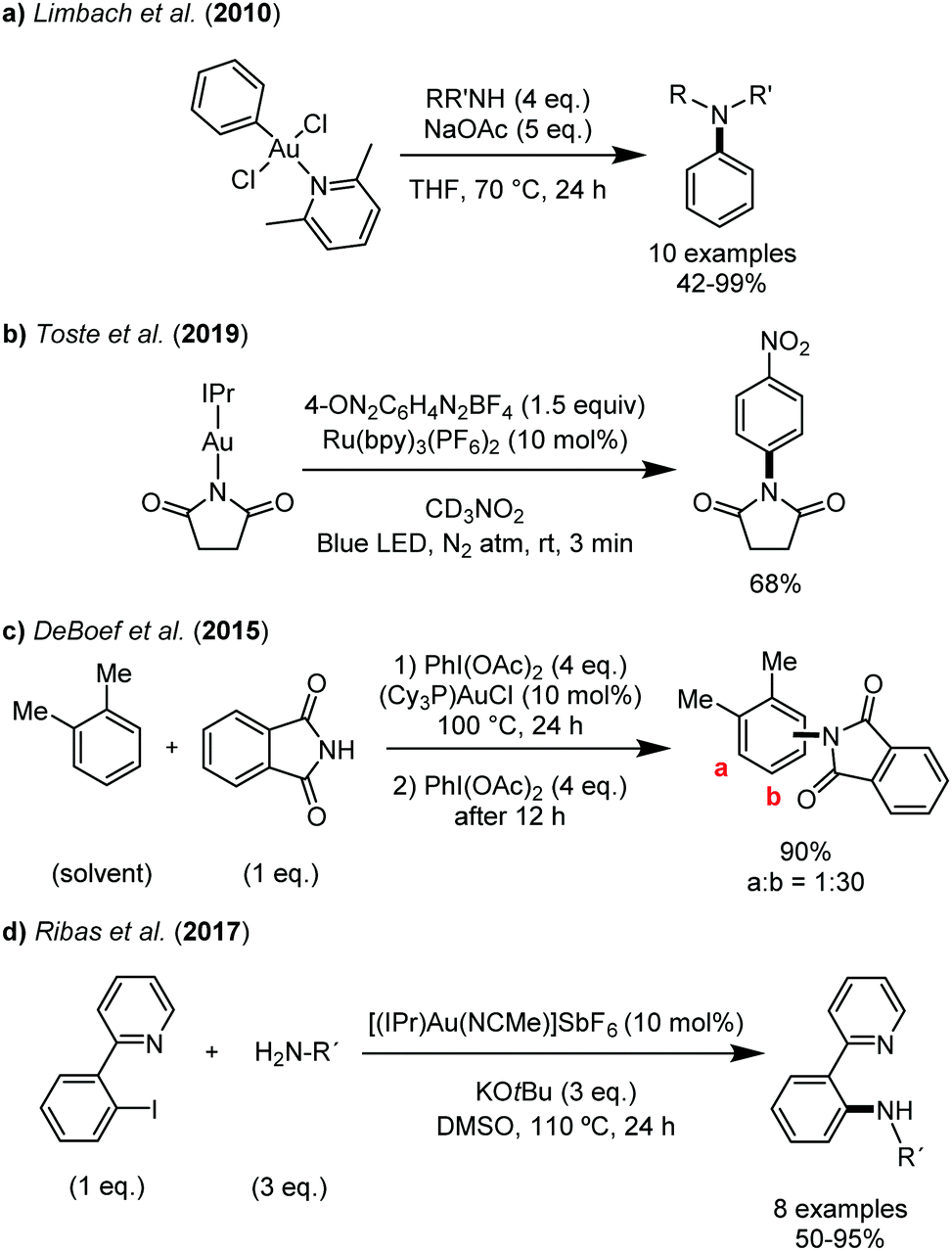 | ||
| Fig. 1 Stoichiometric and catalytic C–N coupling reactions mediated by gold complexes. | ||
With the aim to advance further this chemistry, we have investigated the possibility to catalyze C–N coupling with the (MeDalphos)AuCl complex, taking advantage of the faculty of this gold(I) complex to readily activate aryl iodides. Our results are reported hereafter. The scope and mechanism of the transformation are discussed.17
To start with, the coupling of p-toluenesulfonamide and iodobenzene was used as a benchmark reaction. The influence of different reaction parameters (ratio of the coupling partners, T, halide scavenger, base, solvent) was studied (Table 1).19 The coupling product 1 was obtained in 99% yield using the conditions as given in entry 1. All other conditions resulted in lower yields, such as the use of K3PO4 or NaOAcF as a base, or other halide scavengers such as AgSbF6 or AgBF4 (entries 2–5). The highest yield was obtained using o-DCB/MeOH 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as solvent (entries 6 and 7). Increasing further the temperature was not beneficial (entry 8).
:1 as solvent (entries 6 and 7). Increasing further the temperature was not beneficial (entry 8).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | T (°C) | Halide scavenger | Base | Solventa | Yield (%) | |
| a Solvent/MeOH = 50/1. | ||||||
| 1 | 75 | AgOTf | DTBP | DCB/MeOH | 99 | |
| 2 | 75 | AgOTf | K3PO4 | DCB/MeOH | 58 | |
| 3 | 75 | AgOTf | NaOAcF | DCB/MeOH | 10 | |
| 4 | 75 | AgSbF6 | DTBP | DCB/MeOH | 72 | |
| 5 | 75 | AgBF4 | DTBP | DCB/MeOH | 77 | |
| 6 | 75 | AgOTf | DTBP | DCB | 86 | |
| 7 | 75 | AgOTf | DTBP | DMF/MeOH | 92 | |
| 8 | 100 | AgOTf | DTBP | DCB/MeOH | 80 | |
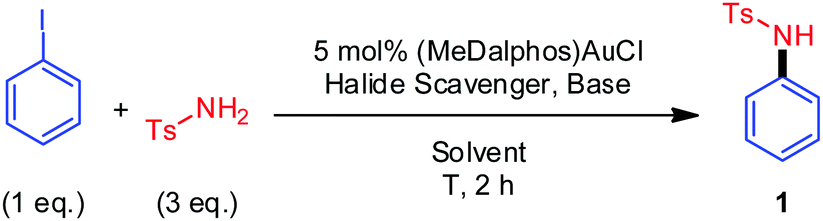
With the optimized conditions in hand, the coupling of 4-iodotoluene with different N-nucleophiles was investigated (Fig. 2a), starting with anilines. Aniline itself gave a very low yield (7%) and no coupling was observed with p-anisidine. Gratifyingly, anilines bearing electron-withdrawing substituents in the para position (NO2, CF3 or COMe) gave the corresponding C–N coupling products 4–6 in good to excellent yields.18 The ortho-substituted 2-methyl-4-nitroaniline also reacted smoothly and cleanly to afford 7. Note that the anilines react selectively at the nitrogen atom (no trace of C-arylated products were detected), whereas indole was found to exclusively undergo C3-arylation under similar conditions.8
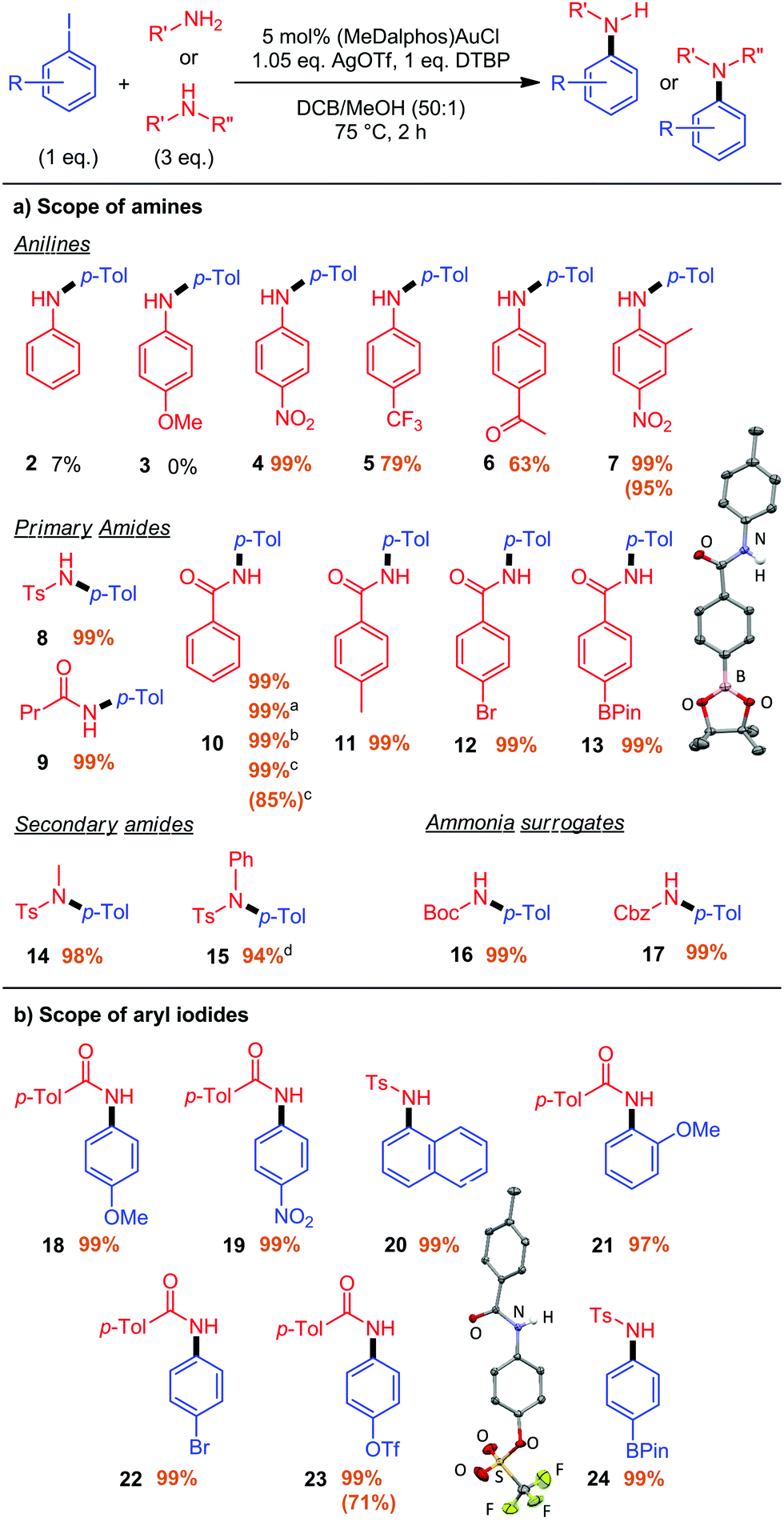 | ||
| Fig. 2 (a) Scope of amines. Cross-coupling of p-iodotoluene and N-nucleophiles catalyzed by (MeDalphos)AuCl at 0.2 M of aryl iodide. aBenzamide (1 eq.), 2 mol% (MeDalphos)AuCl, 4 h, 0.6 M. bAll reagents weighted in air. cBenzamide (2 mmol, 1 eq.), 1 mol% (MeDalphos)AuCl, overnight, 0.6 M. d16 h. (b) Scope of aryl iodides. Cross-coupling of aryl iodides and p-toluenesulfonamide or p-toluamide catalyzed by (MeDalphos)AuCl at 0.1 M of aryl iodide. Yields determined using 1H NMR with n-dodecane as an internal standard. Isolated yields in parentheses. | ||
Besides p-toluenesulfonamide, the reaction works very well with aliphatic and aromatic amides as shown by the quantitative formation of products 9–13. Importantly, the presence of functional groups is compatible with the gold-catalyzed arylation, as shown by the high-yield preparation of Br- and Bpin-substituted N-aryl amides 12 and 13. The structure of 13 was unambiguously confirmed by X-ray diffraction analysis.19 Selective activation of aryl iodides in the presence of Ar–Br and Ar–BPin bonds is particularly noteworthy as it raises selectivity issues with Pd catalysts.
In all cases, the C–N coupling of the primary aromatic amines and amides is highly selective in monoarylation and diarylation products were not detected. Nevertheless, the gold catalyst also promotes the arylation of secondary amides as exemplified by the formation of 14–15. The synthesis of 15 is particularly noteworthy. Starting from p-toluenesulfonamide, compound 1 can be obtained by C–N coupling with iodobenzene and further arylated with 4-iodotoluene using extended reaction times. Thereby, it is possible to sequentially introduce two different aryl groups at nitrogen.
The preparation of primary anilines in protected forms is also possible and straightforward using tert-butyl carbamate (Boc) and benzyl carbamate (Cbz) as ammonia surrogates. The corresponding products 16 and 17 were readily obtained in quantitative yields.
The robustness and scalability of the gold-catalyzed reaction was assessed using 4-iodotoluene and benzamide as substrates. The corresponding product 10 was also obtained in excellent yield in air and with technical grade solvents, showing that no stringent precautions are needed. Very good results were obtained at larger scale. Starting from 2 mmol of substrates with only 1 eq. of benzamide and 1 mol% of (MeDalphos)AuCl, the arylated product 10 was obtained in 99% yield after overnight reaction.
The scope of aryl iodides was then assessed (Fig. 2b). The reaction works well with electron-rich, electron-poor and ortho-substituted substrates, as shown by the quantitative formation of 18–21. Complete selectivity for C–I bond activation in the presence of C–Br, C–OTf and C–Bpin functional groups is observed upon synthesis of 22–24 (the structure of 23 was confirmed by X-ray diffraction analysis),19 highlighting the complementarity and orthogonality of gold and palladium catalysis.
Having established the efficiency and generality of the gold-catalyzed C–N coupling, we sought to gain insight into the reaction mechanism. As schematically depicted in Fig. 3a, two main pathways can be envisioned. They differ essentially in the order of the transmetalation/oxidative addition steps.
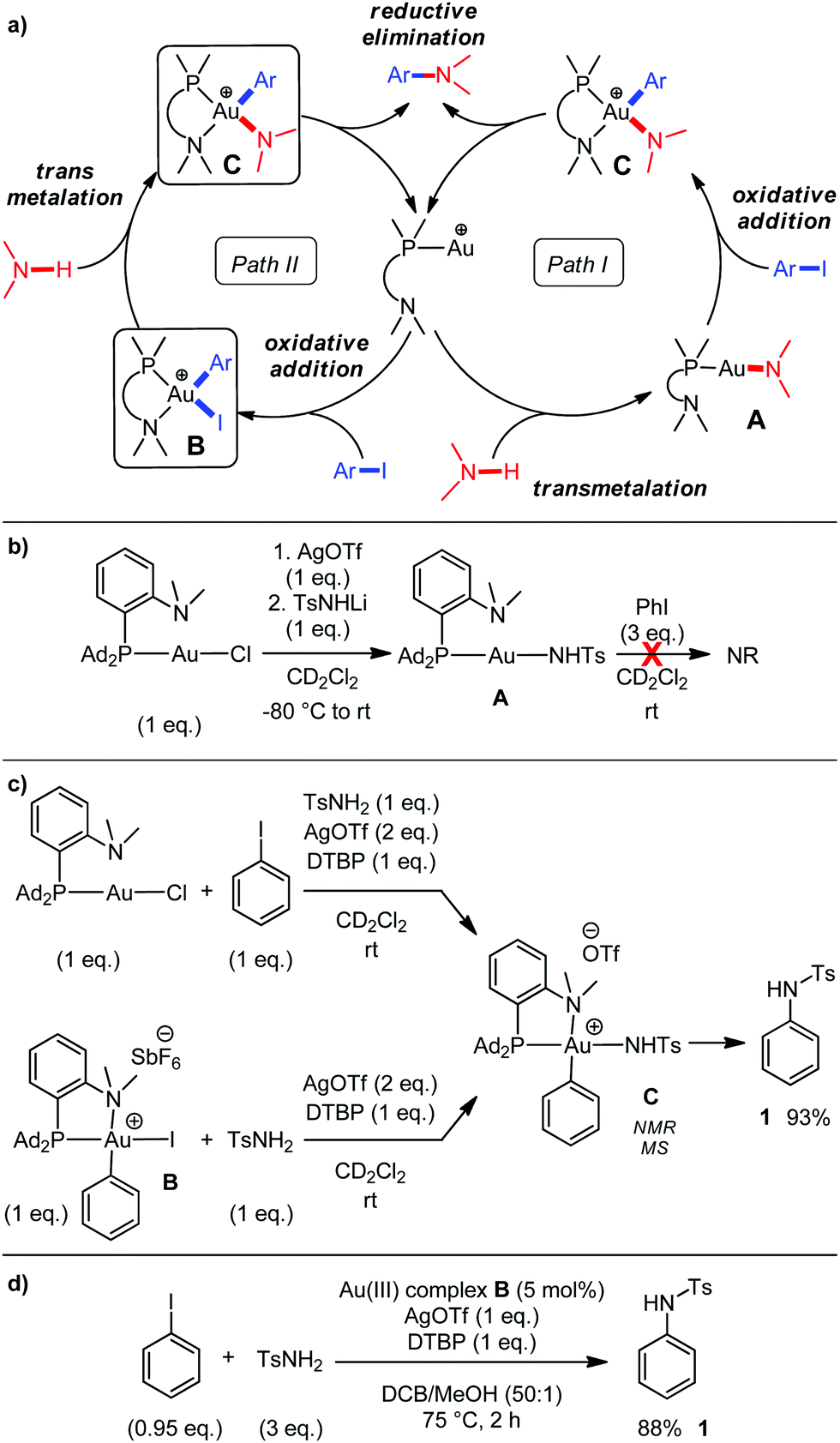 | ||
| Fig. 3 Catalytic cycles envisioned to account for the C–N coupling mediated by bond formation operating via aryl-Au(III) intermediates (a), stoichiometric and catalytic reactions carried out to gain mechanistic insight (b–d). Yields of 1 were determined by 1H NMR with n-dodecane as an internal standard. | ||
Toste et al. have shown that in the gold-promoted C–N coupling of succinimide and aryl diazoniums under photoredox conditions, transmetalation precedes aryl transfer to gold (path I).13 To assess the feasibility of this route with our system, the amido Au(I) complex A was prepared (Fig. 3b). Its structure was analyzed by multi-nuclear NMR. Of note, the 1H–15N HSQC NMR spectrum shows 15N NMR signals at δ 38.1 ppm for the NMe2 group (close to that of the (MeDalphos)AuCl complex, δ 37.0 ppm) and δ 110.2 ppm for the NHTs group with a JNP coupling constant of 37 Hz. X-ray diffraction data for the 2LiOTf adduct was also obtained.19 Despite the presence of the (P,N) ligand to favor oxidative addition and stabilize the ensuing Au(III) species,7,8,20 complex A was found to be completely inert towards iodobenzene (no sign of a reaction after 3 days at room temperature in the presence of 3 eq. of PhI).19
From this observation, we surmised that oxidative addition precedes transmetalation in our case (path II). To support this hypothesis, we studied the influence of N-nucleophiles as additives on the oxidative addition of iodobenzene to the (MeDalphos)AuCl complex.21 No significant impact was noticed with p-nitroaniline and p-toluenesulfonamide, and complex B forms very rapidly and quantitatively. Conversely, the presence of either aniline or p-anisidine slows down the reaction and the oxidative addition is not complete even after 5 h, in line with the absence or very low catalytic activity observed with those substrates.19
Next, we sought to characterize reactive intermediates by running a coupling reaction at rt using 1 eq. of (MeDalphos)AuCl (Fig. 3c). 31P NMR monitoring showed fast and clean formation of a new product at δ 62 ppm. 1H–15N HSQC experiments allowed this species to be identified as the aryl amido Au(III) complex C. The 15N NMR signal for the NMe2 now appears at δ 72.8 ppm, in the typical range of (P,N)-chelated Au(III) complexes (δ 71.5 ppm for complex B) while the NHTs group resonates at δ 117.8 ppm as a doublet. The value of the JNP coupling constant (36 Hz, very similar to that observed for complex A) supports trans arrangement of the amido group and phosphorus atom at gold. The structure of C was further confirmed by high resolution electrospray ionization mass spectrometry (m/z = 865.3232).19 Complex C slowly evolves at rt to regenerate the (MeDalphos)Au(I)+ complex and release the cross-coupling product 1 (93% yield after 36 h).22 The aryl Au(III) complex B is not observed during the course of the reaction, but its involvement is supported by the following reactions performed starting from isolated samples of B: (i) under stoichiometric conditions at rt, it also gives complex C that slowly undergoes reductive elimination and releases the C–N coupling product (Fig. 3c), (ii) under the optimized catalytic reactions, its activity is very similar to that of the (MeDalphos)Au(I)+ complex (Fig. 3d).
Thus, all observations are consistent and support the 2e redox cycle II for this gold-catalyzed C–N coupling. Oxidative addition of the aryl iodide precedes transmetalation and reductive elimination is the rate-determining step.
In summary, we have developed a new C–N coupling methodology that proceeds via a Au(I)/Au(III) catalytic cycle and draws on the faculty of the (P,N) ligand MeDalphos to promote oxidative addition. The reaction does not require the presence of an external oxidant or a directing group. It is robust, operates under mild conditions and works well with a variety of aryl iodides and amines. Most noticeable is the tolerance of functional groups that tend to raise selectivity issues with the other transition metals. Mechanistic studies, including the characterization of a key Au(III) intermediate, strongly support a catalytic sequence made of oxidative addition, transmetalation and rate-determining reductive elimination.
Financial support from the Centre National de la Recherche Scientifique and the Université de Toulouse is gratefully acknowledged. J. R. thanks the European Commission for an MCIF (Gold3Cat-799606). The NMR service of ICT is acknowledged for assistance with the 1H–15N HSQC NMR experiments.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1994–2015 CrossRef CAS PubMed.
- (a) G. Zhang, Y. Peng, L. Cui and L. Zhang, Angew. Chem., Int. Ed., 2009, 48, 3112–3115 CrossRef CAS PubMed; (b) M. N. Hopkinson, A. Tessier, A. Salisbury, G. T. Giuffredi, L. E. Combettes, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2010, 16, 4739–4743 CrossRef CAS PubMed; (c) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644–1648 CrossRef CAS; (d) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, J. Am. Chem. Soc., 2014, 136, 254–264 CrossRef CAS; (e) X. C. Cambeiro, N. Ahlsten and I. Larrosa, J. Am. Chem. Soc., 2015, 137, 15636–15639 CrossRef CAS; (f) L. Huang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 4808–4813 CrossRef CAS PubMed; (g) T. J. A. Corrie, L. T. Ball, C. A. Russell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 245–254 CrossRef CAS; (h) M. Hofer, A. Genoux, R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1021–1025 CrossRef CAS.
- For reviews, see: (a) P. Garcia, M. Malacria, C. Aubert, V. Gandon and L. Fensterbank, ChemCatChem, 2010, 2, 493–497 CrossRef CAS; (b) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2011, 17, 8248–8262 CrossRef CAS; (c) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236–8247 CrossRef CAS; (d) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910–1925 RSC; (e) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS; (f) M. O. Akram, S. Banerjee, S. S. Saswade, V. Bedi and N. T. Patil, Chem. Commun., 2018, 54, 11069–11083 RSC; (g) M. Zidan, S. Rohe, T. McCalluma and L. Barriault, Catal. Sci. Technol., 2018, 8, 6019–6028 RSC; (h) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS.
- (a) M. Schuler, F. Silva, C. Bobbio, A. Tessier and V. Gouverneur, Angew. Chem., Int. Ed., 2008, 47, 7927–7930 CrossRef CAS; (b) M. Hofer and C. Nevado, Tetrahedron, 2013, 69, 5751–5757 CrossRef CAS.
- M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022–15045 CrossRef CAS.
- 2,2′-Bipyridine ligands were also shown to promote oxidative addition to gold and subsequent reaction with an aryl zinc reagent induced C–C cross-coupling (stoichiometric Negishi-type reaction): M. J. Harper, C. J. Arthur, J. Crosby, E. J. Emmett, R. L. Falconer, A. J. Fensham-Smith, P. J. Gates, T. Leman, J. E. McGrady, J. F. Bower and C. A. Russell, J. Am. Chem. Soc., 2018, 140, 4440–4445 CrossRef CAS . For recent insights on ligand influence, see: Y. Yang, L. Eberle, F. F. Mulks, J. F. Wunsch, M. Zimmer, F. Rominger, M. Rudolph and A. S. K. Hashmi, J. Am. Chem. Soc., 2019, 141, 17414–17420 CrossRef.
- A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565–572 CrossRef.
- J. Rodríguez, A. Zeineddine, E. D. Sosa-Carrizo, K. Miqueu, N. Saffon-Merceron, A. Amgoune and D. Bourissou, Chem. Sci., 2019, 10, 7183–7192 RSC.
- (a) M. S. Messina, J. M. Stauber, M. A. Waddington, A. L. Rheingold, H. D. Maynard and A. M. Spokoyny, J. Am. Chem. Soc., 2018, 140, 7065–7069 CrossRef CAS; (b) J. M. Stauber, E. A. Qian, Y. Han, A. L. Rheingold, P. Král, D. Fujita and A. M. Spokoyny, ChemRxiv, 2019 DOI:10.26434/chemrxiv.9783800.
- (a) A. S. Guram and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 7901–7902 CrossRef CAS; (b) F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969–5970 CrossRef CAS.
- (a) D. M. T. Chan, K. L. Monaco, R.-P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef CAS; (b) P. Y. S. Lam, C. G. Clark, S. Saubern, J. Adams, M. P. Winters, D. M. T. Chan and A. Combs, Tetrahedron Lett., 1998, 39, 2941–2944 CrossRef CAS; (c) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS; (d) F. W. Patureau and L. J. Gooßen, Angew. Chem., Int. Ed., 2014, 53, 5738–5739 CrossRef CAS.
- S. Lavy, J. J. Miller, M. Pažický, A.-S. Rodrigues, F. Rominger, C. Jäkel, D. Serra, N. Vinokurov and M. Limbach, Adv. Synth. Catal., 2010, 352, 2993–3000 CrossRef CAS.
- S. Kim and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 4308–4315 CrossRef CAS.
- L. Marchetti, A. Kantak, R. Davis and B. DeBoef, Org. Lett., 2015, 17, 358–361 CrossRef CAS.
- J. Serra, T. Parella and X. Ribas, Chem. Sci., 2017, 8, 946–952 RSC.
- For a stoichiometric intramolecular C–N coupling from a C^N-cyclometalated Au(III) complex, see: J. H. Kim, R. T. Mertens, A. Agarwal, S. Parkin, G. Berger and S. G. Awuah, Dalton Trans., 2019, 48, 6273–6282 RSC.
- During the final stage of this manuscript preparation, a related work by Patil and co-workers was published on line: M. O. Akram, A. Das, I. Chakrabarty and N. T. Patil, Org. Lett., 2019, 21, 8101–8105 CrossRef CAS.
- A similar trend was observed by Ribas et al.15.
- See ESI† for details.
- L. Huang, F. Rominger, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2016, 52, 6435–6438 RSC.
- For recent discussions on the assessment of the functional group tolerance of chemical reactions, see: (a) K. D. Collins and F. Glorius, Acc. Chem. Res., 2015, 48, 619–627 CrossRef CAS PubMed; (b) L. Pitzer, F. Schäfers and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 8572–8576 CrossRef CAS.
- Complete conversion of C takes 0.5 h at 75 °C.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, analytical data and NMR spectra. Crystallographic data for 13, 23 and A (CIF). CCDC 1956358–1956360. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc07666b |
| This journal is © The Royal Society of Chemistry 2020 |