
Regioselective C–H amination of free base porphyrins via electrogenerated pyridinium-porphyrins and stabilization of easily oxidized amino-porphyrins by protonation†

Abstract
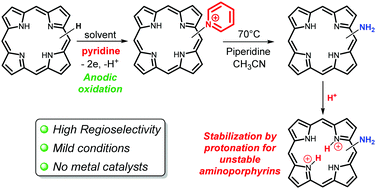
Four free base aminoporphyrins were synthesized in two steps via regioselective anodic nucleophilic substitution with pyridine followed by ring opening of the electrogenerated pyridinium with piperidine. The X-ray crystallographic structure of the unstable 2-aminotetraphenylporphyrin was solved. Protonation of this latter compound leads to the stable diiminium porphyrin salt.
 Please wait while we load your content...
Please wait while we load your content...

