
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUV-induced hydrogen transfer in DNA base pairs promoted by dark nπ* states†
Kinga E.
Szkaradek
a,
Petr
Stadlbauer
bc,
Jiří
Šponer
 bc,
Robert W.
Góra
*a and
Rafał
Szabla
*cd
bc,
Robert W.
Góra
*a and
Rafał
Szabla
*cd
aDepartment of Physical and Quantum Chemistry, Wroclaw University of Science and Technology, Faculty of Chemistry, Wybrzeże Wyspiańskiego 27, 50-370, Wrocław, Poland. E-mail: robert.gora@pwr.edu.pl
bRegional Centre of Advanced Technologies and Materials, Faculty of Science, Palacky University, Šlechtitelů 27, 771 46 Olomouc, Czech Republic
cInstitute of Biophysics of the Czech Academy of Sciences, Královopolská 135, 61265 Brno, Czech Republic
dInstitute of Physics, Polish Academy of Sciences, Al. Lotników 32/46, 02-668 Warsaw, Poland. E-mail: szabla@ifpan.edu.pl
First published on 4th December 2019
Abstract
Dark nπ* states were shown to have substantial contribution to the destructive photochemistry of pyrimidine nucleobases. Based on quantum-chemical calculations, we demonstrate that the characteristic hydrogen bonding pattern of the GC base pair could facilitate the formation of a wobble excited-state charge-transfer 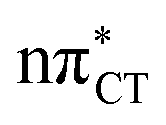 complex. This entails a barrierless electron-driven proton transfer (EDPT) process which enables damageless photodeactivation of the base pair. These photostabilizing properties are retained even when guanine is exchanged to hypoxanthine. The inaccessibility of this process in the AT base pair sheds further light on the reasons why cytosine is less susceptible to the formation of photodimers in double-stranded DNA.
complex. This entails a barrierless electron-driven proton transfer (EDPT) process which enables damageless photodeactivation of the base pair. These photostabilizing properties are retained even when guanine is exchanged to hypoxanthine. The inaccessibility of this process in the AT base pair sheds further light on the reasons why cytosine is less susceptible to the formation of photodimers in double-stranded DNA.
Photoinduced electron transfer is a ubiquitous phenomenon that could trigger various photochemical reactions, regulates the efficiency of organic solar cells and contributes to the photophysics and repair of DNA.1 Such charge migration in biomolecules often entails a proton transfer and subsequent efficient photodeactivation through a crossing between the S1 and S0 states.2 This process is often referred to as electron-driven proton transfer (EDPT)3 or sequential proton-coupled electron transfer (PCET)4 and was suggested as an origin of short excited-state lifetimes and photostability of the key biomolecular building blocks5 and microsolvated organic chromophores.6 One of the most representative examples of EDPT was reported for the Watson–Crick (WC) guanine–cytosine (GC) base pair in the gas phase, based on pump–probe spectroscopic measurements and quantum chemical calculations.7 These findings enabled to assign the accessibility of the EDPT mechanism in WC base pairs to the presence of low-energy
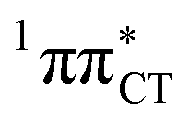 charge-transfer (CT) states, involving electronic transitions between bonding π molecular orbital localized on guanine and antibonding π* orbital localized on cytosine (cf.Fig. 2). More recent studies demonstrated that EDPT could also contribute to the photostability of GC in solution and in the native environment of DNA double helix.8
charge-transfer (CT) states, involving electronic transitions between bonding π molecular orbital localized on guanine and antibonding π* orbital localized on cytosine (cf.Fig. 2). More recent studies demonstrated that EDPT could also contribute to the photostability of GC in solution and in the native environment of DNA double helix.8
In opposition to the photostabilizing EDPT mechanism, pyrimidine nucleosides were shown to follow a competing photodeactivation pathway associated with the population of locally excited (LE) 1nπ* states with lifetimes exceeding tens of ps.9 These dark states could further serve as a doorway to even longer-lived and highly-reactive triplet 3ππ* electronic states10 enabling dimerization of adjacent pyrimidine bases.11 The 1nOπ* state accessed after photoexcitation of cytidine was also demonstrated to enable abstraction of the C1′–H hydrogen atom from the sugar moiety and subsequent photoanomerisation from the naturally occurring β to the α anomer.12 Finally, 1nπ* states were suggested to participate in a water-splitting reaction in which the nucleobase abstracts a hydrogen atom from the neighboring water molecule and generates a hydroxyl radical.13 The ˙OH radical could further attack the hydrogenated chromophore radical leading to the formation of a photohydrate, yet another type of photolesion that could significantly impede the functions of DNA and RNA.13
Despite similar contribution of 1nπ* states to the photodynamics of the three canonical pyrimidine RNA/DNA bases,9 cytosine was found to be the least susceptible to form photolesions in nucleic acid strands.14 Therefore, our working hypothesis is that the lowest-energy 1nπ* state of cytosine might promote an efficient photorelaxation channel which is unavailable in the AT and AU WC base pairs. To verify this hypothesis, we investigated the GC base pair focusing on possible electron transfer processes occurring on the 1nπ* hypersurface and the associated state crossings. We also considered the WC base pair containing the alternative nucleobase hypoxanthine as a substitute of guanine (HC base pair). Hypoxanthine nucleoside (inosine) was recently suggested to be a potential component of primordial versions of RNA.15 Thus the HC base pair could serve as a valuable reference point to understand the photochemical processes in the GC base pair. We performed explorations of the excited-state potential-energy (PE) surfaces of these two base pairs using the algebraic diagrammatic construction to the second order [ADC(2)] method as implemented in the Turbomole 7.3 package.16
We started our study with optimizing the ground state geometries of the GC and HC WC base pairs in the gas phase using the MP2/cc-pVTZ method (see Fig. 1). To separate 1ππ* and 1nπ* excitations we assumed the Cs point-group symmetry corresponding to planar structures. However, analogous optimizations of these base pairs performed without any constraints returned nearly planar geometries as well. Despite only two interbase hydrogen bonds the HC base pair is characterized by a virtually identical orientation of the interacting bases to its biological counterpart GC. Slight differences in the common structural motifs are thus the result of the absence of the exocyclic amino group in hypoxanthine and the absence of N–H⋯O hydrogen bond in HC.
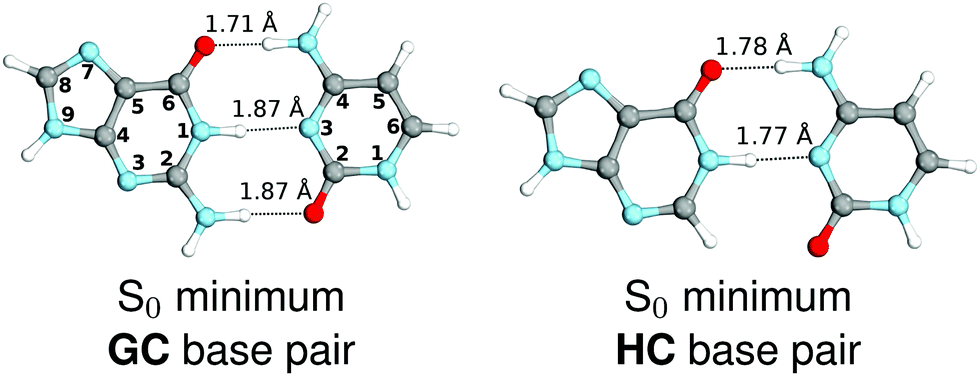 | ||
| Fig. 1 Ground-state minimum-energy geometries of the HC and GC base pairs optimized at the MP2/cc-pVTZ level of theory. | ||
The above structural similarities are also reflected by the vertical excitation energies of the LE states presented in Table 1 (obtained at the ADC(2)/cc-pVTZ level). Both HC and GC are characterized by highly consistent energies and oscillator strengths of low-lying bright ππ* states, what indicates very similar spectral properties of the two WC base pairs. However, the low-lying  state, characterized by an electron transferred from purine to pyrimidine, is significantly destabilized in HC (5.91 eV) when compared to GC (5.16 eV). This state was attributed to the ultrafast electron-driven proton transfer (EDPT) photodeactivation mechanism in GC7 and we expect that the EDPT process may be unavailable in HC at lower excitation energies. This is further supported by the presence of low-lying 1nπ* states in the spectrum, due to dipole-forbidden transitions from nonbonding electron pair to π* orbital, which could have a significant contribution to the photorelaxation of the HC base pair.
state, characterized by an electron transferred from purine to pyrimidine, is significantly destabilized in HC (5.91 eV) when compared to GC (5.16 eV). This state was attributed to the ultrafast electron-driven proton transfer (EDPT) photodeactivation mechanism in GC7 and we expect that the EDPT process may be unavailable in HC at lower excitation energies. This is further supported by the presence of low-lying 1nπ* states in the spectrum, due to dipole-forbidden transitions from nonbonding electron pair to π* orbital, which could have a significant contribution to the photorelaxation of the HC base pair.
| State/transition | E exc/[eV] | f osc | λ/[nm] | |
|---|---|---|---|---|
| GC C S symmetry | ||||
| S1(A′) | ππ* | 4.86 | 6.99 × 10−2 | 255.3 |
| S2(A′) | ππ* | 4.91 | 5.65 × 10−2 | 252.7 |
| S3(A′) |
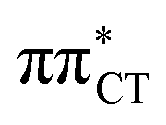
|
5.16 | 2.84 × 10−2 | 240.1 |
| S4(A′′) |
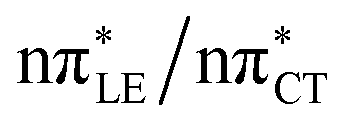
|
5.37 | 6.45 × 10−4 | 230.9 |
| S5(A′) | ππ* | 5.37 | 0.234 | 230.9 |
| S6(A′) | ππ* | 5.42 | 0.407 | 228.9 |
| HC C S symmetry | ||||
| S1(A′) | ππ* | 4.809 | 7.60 × 10−2 | 257.8 |
| S2(A′) | ππ* | 5.044 | 8.70 × 10−2 | 245.8 |
| S3(A′′) |
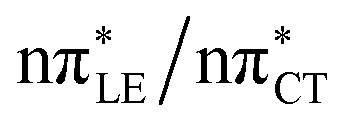
|
5.227 | 2.97 × 10−4 | 237.2 |
| S4(A′) | ππ* | 5.344 | 0.236 | 232.0 |
| S5(A′′) | nπ* | 5.469 | 4.24 × 10−4 | 226.7 |
| S10(A′) |
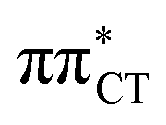
|
5.912 | 4.31 × 10−3 | 209.7 |
Based on this initial analysis of vertical excitations it is reasonable to infer that HC might indeed be much more vulnerable to photodamage than GC. However, we observed one additional and elusive feature of the reactive 1nπ* excitations which could have intriguing consequences for the photodeactivation of both considered base pairs. In each case, the lowest energy 1nπ* states exhibit partial charge-transfer (CT) character that could be the driving force for yet another deactivation mechanism that was not reported previously. This partial 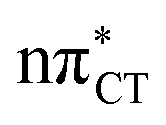 character is indicated by 0.17 of e− charge transferred from carbonyl lone-pair of G to π* orbital localized on C and 0.08 of e− charge transferred from H to C in the Franck–Condon regions of these base pairs (i.e. at their ground-state geometries).
character is indicated by 0.17 of e− charge transferred from carbonyl lone-pair of G to π* orbital localized on C and 0.08 of e− charge transferred from H to C in the Franck–Condon regions of these base pairs (i.e. at their ground-state geometries).
To further investigate the photoreactivity of the 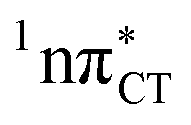 states, we performed optimizations of S1 minima, again imposing planar (Cs) symmetry restrictions. The 1nπ* state with partial CT character is thus the lowest excited singlet state belonging to the A′′ irreducible representation in both GC and HC base pairs. The corresponding optimized geometries associated with the plateau region on the
states, we performed optimizations of S1 minima, again imposing planar (Cs) symmetry restrictions. The 1nπ* state with partial CT character is thus the lowest excited singlet state belonging to the A′′ irreducible representation in both GC and HC base pairs. The corresponding optimized geometries associated with the plateau region on the 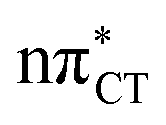 surface are characterized by a substantial increase of the CT character up to 0.57 and 0.52 of e− for the GC and HC base pairs, respectively. This partial electron transfer from the purine to the pyrimidine base may also result in a subsequent proton transfer process. In fact, this EDPT mechanism is enabled by significant displacement of the two nucleobases and formation of an excited-state complex (exciplex). The corresponding geometry of the S1(
surface are characterized by a substantial increase of the CT character up to 0.57 and 0.52 of e− for the GC and HC base pairs, respectively. This partial electron transfer from the purine to the pyrimidine base may also result in a subsequent proton transfer process. In fact, this EDPT mechanism is enabled by significant displacement of the two nucleobases and formation of an excited-state complex (exciplex). The corresponding geometry of the S1(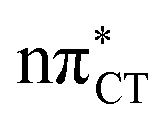 ) PE minimum can be described as a wobble GC base pair with a hydrogen atom transferred photochemically from G to C (see Fig. 2). Formation of the excited-state wobble base pair is supported by one hydrogen bond and a N⋯O interaction in which the electron-deficient carbonyl nO molecular orbital of G borrows electron density from the nN molecular orbital of C. The corresponding N⋯O distance amounts to 2.06 and 2.07 Å in HC and GC, respectively. The N⋯O interaction is the key structural feature that enhances charge transfer character of the lowest energy nπ* state outside the Franck–Condon region and enables subsequent proton transfer (compare Fig. 2 and 1). Finally, the energy gap separating the S1 and S0 states at the S1(
) PE minimum can be described as a wobble GC base pair with a hydrogen atom transferred photochemically from G to C (see Fig. 2). Formation of the excited-state wobble base pair is supported by one hydrogen bond and a N⋯O interaction in which the electron-deficient carbonyl nO molecular orbital of G borrows electron density from the nN molecular orbital of C. The corresponding N⋯O distance amounts to 2.06 and 2.07 Å in HC and GC, respectively. The N⋯O interaction is the key structural feature that enhances charge transfer character of the lowest energy nπ* state outside the Franck–Condon region and enables subsequent proton transfer (compare Fig. 2 and 1). Finally, the energy gap separating the S1 and S0 states at the S1(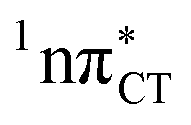 ) minima drops below 0.6 eV which indicates that the associated S1/S0 state crossings are nearly reached.
) minima drops below 0.6 eV which indicates that the associated S1/S0 state crossings are nearly reached.
 | ||
Fig. 2 Minimum-energy geometries of the 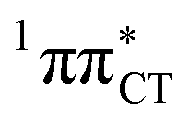 and and 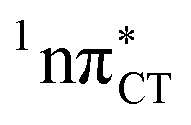 states and the associated occupied (solid blue and violet) and virtual (translucent yellow and green) molecular orbitals. states and the associated occupied (solid blue and violet) and virtual (translucent yellow and green) molecular orbitals. | ||
The PE profile corresponding to the formation of the wobble geometry and the subsequent EDPT process in GC is presented in Fig. 3. This PE profile demonstrates that the wobble exciplex geometry of GC can be formed in a barrierless manner upon the population of the lowest energy 1nπ* state. This in-plane dislocation of the two bases results in a complete electron transfer from G to C and drives the base pair towards a plateau on the S1 PE surface. The subsequent proton transfer may occur on this plateau and enable the formation of the S1/S0 state crossing (conical intersection). Therefore, the EDPT process occurring on the 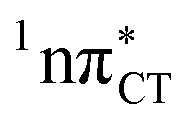 hypersurface should be considered as a two-stage mechanism, as opposed to one-stage EDPT driven by the
hypersurface should be considered as a two-stage mechanism, as opposed to one-stage EDPT driven by the  state, which was described by Sobolewski and co-workers (see the inset of Fig. 3).7 The results of our ADC(2) calculations were additionally benchmarked against the SCS-ADC(2) approach and higher level XMS-CASPT2 computations (Section 2.4 in the ESI†). We have also calculated the PE profiles for these two distinct EDPT processes in HC (Section 2.3 in the ESI†). The two-stage EDPT process occurring on the
state, which was described by Sobolewski and co-workers (see the inset of Fig. 3).7 The results of our ADC(2) calculations were additionally benchmarked against the SCS-ADC(2) approach and higher level XMS-CASPT2 computations (Section 2.4 in the ESI†). We have also calculated the PE profiles for these two distinct EDPT processes in HC (Section 2.3 in the ESI†). The two-stage EDPT process occurring on the 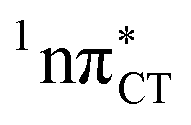 surface of HC is virtually identical to what we have already described for GC. However, owing to very high excitation energy of the
surface of HC is virtually identical to what we have already described for GC. However, owing to very high excitation energy of the 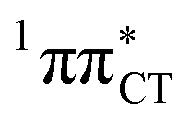 state of HC (5.91 eV) the one-stage EDPT process in this base pair could only be triggered at substantially higher excitation energies than in the case of GC.
state of HC (5.91 eV) the one-stage EDPT process in this base pair could only be triggered at substantially higher excitation energies than in the case of GC.
 | ||
Fig. 3 PE surface cuts illustrating the EDPT mechanism driven by the 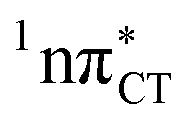 state. The PE profile was constructed as a linear interpolation in internal coordinates (LIIC) between the FC region, S1 plateau and the state. The PE profile was constructed as a linear interpolation in internal coordinates (LIIC) between the FC region, S1 plateau and the 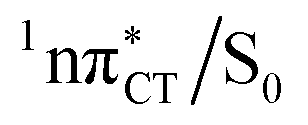 state crossing. The x axis in the first part of the profile corresponds to the O⋯N3 distance, characteristic for the exciplex interaction. The reaction coordinate on the right hand side describes the proton transfer process. The inset shows the one-stage EDPT mechanism triggered on the state crossing. The x axis in the first part of the profile corresponds to the O⋯N3 distance, characteristic for the exciplex interaction. The reaction coordinate on the right hand side describes the proton transfer process. The inset shows the one-stage EDPT mechanism triggered on the 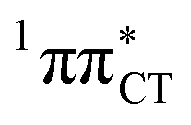 surface. surface. | ||
It is worth noting that similar 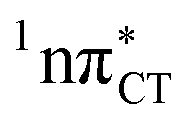 states were also reported in microhydrated cytidine and adenine involving similar state-of-the-art quantum chemical simulations.13 These examples involved partial electron transfer from a neighbouring water molecule to the chromophore moiety and were also suggested to have a significant contribution to the photochemistry of these molecules in water solution.13 In the case of the GC and HC WC base pairs the
states were also reported in microhydrated cytidine and adenine involving similar state-of-the-art quantum chemical simulations.13 These examples involved partial electron transfer from a neighbouring water molecule to the chromophore moiety and were also suggested to have a significant contribution to the photochemistry of these molecules in water solution.13 In the case of the GC and HC WC base pairs the 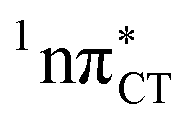 state can serve as a photostabilizing deactivation channel. Consequently, once the
state can serve as a photostabilizing deactivation channel. Consequently, once the 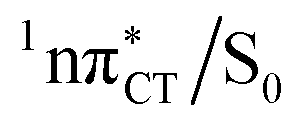 state crossing (shown in Fig. 3) is reached the base pair can repopulate the closed-shell electronic ground-state and the transferred hydrogen atom may be returned to the purine base. This enables barrierless restoration of the canonical structure of the WC base pair.
state crossing (shown in Fig. 3) is reached the base pair can repopulate the closed-shell electronic ground-state and the transferred hydrogen atom may be returned to the purine base. This enables barrierless restoration of the canonical structure of the WC base pair.
The excited-state lifetime of the dark 1nπ* state in aqueous cytidine was established in several independent experiments which consistently returned the value of ∼30 ps.9 The PE surface shown in Fig. 3 demonstrates that when this dark locally-excited nπ* state is populated in GC (or HC) it may easily acquire a CT character. This CT event could in turn significantly shorten the excited-state lifetime by triggering the highly efficient EDPT mechanism. Interestingly, the population of 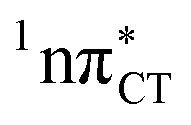 states can be facilitated only in the case of the specific hydrogen bonding pattern present in GC and HC. The formation of an analogous exciplex interaction promoting subsequent proton transfer is not possible in the case of AT and AU pairs. This could explain why both T and U are much more susceptible to photodamage in DNA double strands than C.
states can be facilitated only in the case of the specific hydrogen bonding pattern present in GC and HC. The formation of an analogous exciplex interaction promoting subsequent proton transfer is not possible in the case of AT and AU pairs. This could explain why both T and U are much more susceptible to photodamage in DNA double strands than C.
The specific exciplex interaction associated with the 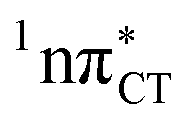 state in GC and HC is structurally similar to the non-native GT wobble pair (see Fig. 4). In each of these cases, the interacting purine and pyrimidine bases are clearly displaced when compared to the native WC pairing pattern, however, crystallographic studies of double-stranded DNA fragments indicate that such displacement should have little or no effect on the sugar-phosphate backbone.17 To examine this problem more closely we performed an MD simulation of the GGGCCC and GGHCCC B-DNA fragments (including the complementary strands) in which we further mutated one of the C bases to a T (see Section 2.5 in the ESI†). We simulated 24 and 12 trajectories for the d(GGGTCC)·(GGGCCC) and d(GGGTCC)·(GGHCCC) systems, respectively, assuming different initial conditions. These simulations revealed that the native B-DNA backbone conformation enables very efficient, and practically ultrafast formation of the wobble GT interaction after the mutation is introduced. This transition was observed during the first 5 ps in 62.5% of all the trajectories initialized for the d(GGGTCC)·(GGGCCC) system and 75% of the trajectories simulated for the d(GGGTCC)·(GGHCCC) system. Therefore, we anticipate that the timescale of the formation of the wobble
state in GC and HC is structurally similar to the non-native GT wobble pair (see Fig. 4). In each of these cases, the interacting purine and pyrimidine bases are clearly displaced when compared to the native WC pairing pattern, however, crystallographic studies of double-stranded DNA fragments indicate that such displacement should have little or no effect on the sugar-phosphate backbone.17 To examine this problem more closely we performed an MD simulation of the GGGCCC and GGHCCC B-DNA fragments (including the complementary strands) in which we further mutated one of the C bases to a T (see Section 2.5 in the ESI†). We simulated 24 and 12 trajectories for the d(GGGTCC)·(GGGCCC) and d(GGGTCC)·(GGHCCC) systems, respectively, assuming different initial conditions. These simulations revealed that the native B-DNA backbone conformation enables very efficient, and practically ultrafast formation of the wobble GT interaction after the mutation is introduced. This transition was observed during the first 5 ps in 62.5% of all the trajectories initialized for the d(GGGTCC)·(GGGCCC) system and 75% of the trajectories simulated for the d(GGGTCC)·(GGHCCC) system. Therefore, we anticipate that the timescale of the formation of the wobble 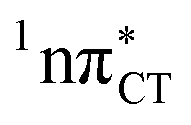 exciplex is likely much shorter than the lifetime of the 1nπ* state in cytidine monomer and this process should be easily accessible for GC and HC in double-stranded DNA.
exciplex is likely much shorter than the lifetime of the 1nπ* state in cytidine monomer and this process should be easily accessible for GC and HC in double-stranded DNA.
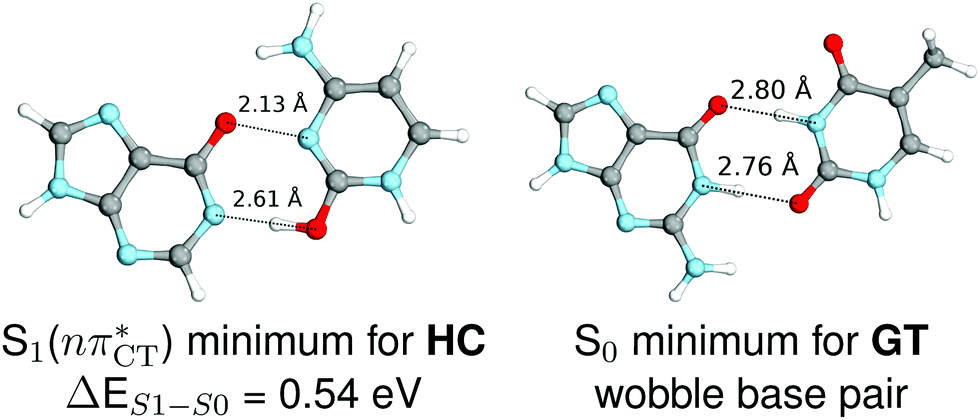 | ||
Fig. 4 Comparison of the S1( ) minimum-energy geometry of HC and the wobble GT base pair in its electronic ground-state. Distances between heteroatoms are marked with dashed lines. ) minimum-energy geometry of HC and the wobble GT base pair in its electronic ground-state. Distances between heteroatoms are marked with dashed lines. | ||
In conclusion, we demonstrated that long-lived and reactive 1nπ* states reported previously as potential sources of photodamage in separate pyrimidine nucleosides may, in fact, facilitate efficient photodeactivation via a two-stage electron-driven proton transfer (EDPT) in specific WC base pairs. This channel is enabled by partial charge-transfer character of the 1nπ* state and the formation of wobble exciplex geometry. In the case of GC, this process could be complementary to the well-documented EDPT photodeactivation mechanism occurring on the 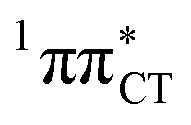 hypersurface.3,7 In contrast, the hydrogen-bonging pattern of AT prohibits the formation of the wobble exciplex structure which could stabilize the
hypersurface.3,7 In contrast, the hydrogen-bonging pattern of AT prohibits the formation of the wobble exciplex structure which could stabilize the 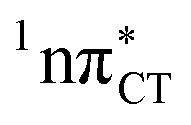 character. This may be yet another explanation why the radiationless deactivation involving EDPT is much more favorable in GC than in AT. Therefore, our findings supplement the rationale behind the substantial photostability of cytosine when compared to thymine and uracil in double-stranded DNA. Our results indicate that this mechanism should be also available in the HC base pair containing the non-canonical nucleobase hypoxanthine, which suggests that HC could undergo ultrafast photorelaxation despite the apparent inaccessibility of EDPT channel triggered on the
character. This may be yet another explanation why the radiationless deactivation involving EDPT is much more favorable in GC than in AT. Therefore, our findings supplement the rationale behind the substantial photostability of cytosine when compared to thymine and uracil in double-stranded DNA. Our results indicate that this mechanism should be also available in the HC base pair containing the non-canonical nucleobase hypoxanthine, which suggests that HC could undergo ultrafast photorelaxation despite the apparent inaccessibility of EDPT channel triggered on the 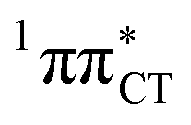 PE surface. Presence of such photostabilizing processes in HC also reinforces the prebiotic scenarios which involve the formation of the hypoxanthine nucleoside inosine and utilize this molecule in enhancing the rate and fidelity of nonenzymatic RNA template copying.15 It is worth emphasizing that the EDPT mechanism promoted by dark
PE surface. Presence of such photostabilizing processes in HC also reinforces the prebiotic scenarios which involve the formation of the hypoxanthine nucleoside inosine and utilize this molecule in enhancing the rate and fidelity of nonenzymatic RNA template copying.15 It is worth emphasizing that the EDPT mechanism promoted by dark  states is likely the only ultrafast photorelaxation mechanism available in HC. While the primary goal of this preliminary account is to supplement the current knowledge about EDPT and the photoreactivity of dark 1nπ* states, more details regarding the role of
states is likely the only ultrafast photorelaxation mechanism available in HC. While the primary goal of this preliminary account is to supplement the current knowledge about EDPT and the photoreactivity of dark 1nπ* states, more details regarding the role of 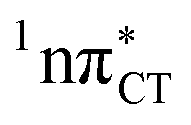 exciplexes in WC base pairs could be revealed in the future by pump–probe experiments that could be performed in supersonic expansions or an apolar solvent mimicking the interior of a DNA helix (e.g. chloroform). HC would be an excellent model system to study this photodeactivation mechanism, since an ultrafast EDPT process triggered in this base pair at lower excitation energies (e.g. ∼4.8 eV) would most likely originate from the
exciplexes in WC base pairs could be revealed in the future by pump–probe experiments that could be performed in supersonic expansions or an apolar solvent mimicking the interior of a DNA helix (e.g. chloroform). HC would be an excellent model system to study this photodeactivation mechanism, since an ultrafast EDPT process triggered in this base pair at lower excitation energies (e.g. ∼4.8 eV) would most likely originate from the 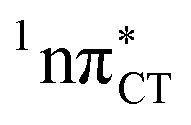 PE surface.
PE surface.
This work was supported in part by a fellowship from the Simons Foundation (494188 to R. S.), by the Foundation for Polish Science (FNP) and a grant from the National Science Centre Poland (2016/23/B/ST4/01048 to R. W. G.). Support from SYMBIT: European Regional Development Fund [CZ.02.1.01/0.0/0.0/15_003/0000477] is gratefully acknowledged. We thank Mikołaj Janicki and Prof. Andrzej Sobolewski for helpful discussions.
Conflicts of interest
There are no conflicts to declare.References
- (a) P. Song, Y. Li, F. Ma, T. Pullerits and M. Sun, Chem. Rev., 2016, 16, 734–753 CAS; (b) C. T. Middleton, K. D. L. Harpe, C. Su, Y. K. Law, C. E. Crespo-Hernández and B. Kohler, Annu. Rev. Phys. Chem., 2009, 60, 217–239 CrossRef CAS PubMed; (c) R. Szabla, H. Kruse, P. Stadlbauer, J. Šponer and A. L. Sobolewski, Chem. Sci., 2018, 9, 3131–3140 RSC; (d) J. J. Nogueira, F. Plasser and L. González, Chem. Sci., 2017, 8, 5682–5691 RSC.
- A. L. Sobolewski and W. Domcke, Europhys. News, 2006, 37, 20–23 CrossRef CAS.
- T. Schultz, E. Samoylova, W. Radloff, I. V. Hertel, A. L. Sobolewski and W. Domcke, Science, 2004, 306, 1765–1768 CrossRef CAS PubMed.
- (a) S. Hammes-Schiffer, Energy Environ. Sci., 2012, 5, 7696–7703 RSC; (b) S. Hammes-Schiffer, J. Am. Chem. Soc., 2015, 137, 8860–8871 CrossRef CAS.
- (a) B. Marchetti, T. N. V. Karsili, M. N. R. Ashfold and W. Domcke, Phys. Chem. Chem. Phys., 2016, 18, 20007–20027 RSC; (b) G. Groenhof, L. V. Schäfer, M. Boggio-Pasqua, M. Goette, H. Grubmüller and M. A. Robb, J. Am. Chem. Soc., 2007, 129, 6812–6819 CrossRef CAS; (c) S. Perun, A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2006, 110, 9031–9038 CrossRef CAS PubMed; (d) K. Röttger, H. J. Marroux, A. F. Chemin, E. Elsdon, T. A. Oliver, S. T. Street, A. S. Henderson, M. C. Galan, A. J. Orr-Ewing and G. M. Roberts, J. Phys. Chem. B, 2017, 121, 4448–4455 CrossRef PubMed; (e) D. Tuna, A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2014, 118, 122–127 CrossRef CAS PubMed; (f) R. Crespo-Otero, A. Mardykov, E. Sanchez-Garcia, W. Sander and M. Barbatti, Phys. Chem. Chem. Phys., 2014, 16, 18877–18887 RSC.
- (a) V. Stert, L. Hesse, H. Lippert, C. Schulz and W. Radloff, J. Phys. Chem. A, 2002, 106, 5051–5053 CrossRef CAS; (b) R. Szabla, J. Šponer and R. W. Góra, J. Phys. Chem. Lett., 2015, 6, 1467–1471 CrossRef CAS; (c) J. J. Nogueira, A. Corani, A. El Nahhas, A. Pezzella, M. d'Ischia, L. González and V. Sundström, J. Phys. Chem. Lett., 2017, 8, 1004–1008 CrossRef CAS PubMed; (d) M. J. Janicki, R. Szabla, J. Šponer and R. W. Góra, Faraday Discuss., 2018, 212, 345–358 RSC; (e) J. Ehrmaier, M. J. Janicki, A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2018, 20, 14420–14430 RSC.
- (a) A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2004, 6, 2763–2771 RSC; (b) A. Abo-Riziq, L. Grace, E. Nir, M. Kabelac, P. Hobza and M. S. D. Vries, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 20–23 CrossRef CAS PubMed; (c) A. L. Sobolewski, W. Domcke and C. Hättig, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17903–17906 CrossRef CAS.
- (a) D. B. Bucher, A. Schlueter, T. Carell and W. Zinth, Angew. Chem., Int. Ed., 2014, 53, 11366–11369 CrossRef CAS; (b) K. Röttger, H. J. Marroux, M. P. Grubb, P. M. Coulter, H. Böhnke, A. S. Henderson, M. C. Galan, F. Temps, A. J. Orr-Ewing and G. M. Roberts, Angew. Chem., Int. Ed., 2015, 54, 14719–14722 CrossRef; (c) A. Francés-Monerris, H. Gattuso, D. Roca-Sanjuán, I. Tuñón, M. Marazzi, E. Dumont and A. Monari, Chem. Sci., 2018, 9, 7902–7911 RSC.
- (a) P. M. Hare, C. E. Crespo-Hernández and B. Kohler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 435–440 CrossRef CAS; (b) P. M. Keane, M. Wojdyla, G. W. Doorley, G. W. Watson, I. P. Clark, G. M. Greetham, A. W. Parker, M. Towrie, J. M. Kelly and S. J. Quinn, J. Am. Chem. Soc., 2011, 133, 4212–4215 CrossRef CAS PubMed; (c) C. Ma, C. C.-W. Cheng, C. T.-L. Chan, R. C.-T. Chan and W.-M. Kwok, Phys. Chem. Chem. Phys., 2015, 17, 19045–19057 RSC; (d) A. J. Pepino, J. Segarra-Mart, A. Nenov, I. Rivalta, R. Improta and M. Garavelli, Phys. Chem. Chem. Phys., 2018, 20, 6877–6890 RSC.
- C. Salet, R. Bensasson and R. Becker, Photochem. Photobiol., 1979, 30, 325–329 CrossRef CAS.
- L. Liu, B. M. Pilles, J. Gontcharov, D. B. Bucher and W. Zinth, J. Phys. Chem. B, 2016, 120, 292–298 CrossRef CAS.
- R. Szabla, J. Campos, J. E. Šponer, J. Šponer, R. W. Góra and J. D. Sutherland, Chem. Sci., 2015, 6, 2035–2043 RSC.
- (a) R. Szabla, H. Kruse, J. Šponer and R. W. Góra, Phys. Chem. Chem. Phys., 2017, 19, 17531–17537 RSC; (b) X. Wu, T. N. V. Karsili and W. Domcke, ChemPhysChem, 2016, 17, 1298–1304 CrossRef CAS.
- S. Mouret, C. Baudouin, M. Charveron, A. Favier, J. Cadet and T. Douki, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13765–13770 CrossRef CAS.
- (a) S. J. Roberts, R. Szabla, Z. R. Todd, S. Stairs, D.-K. Bučar, J. Šponer, D. D. Sasselov and M. W. Powner, Nat. Commun., 2018, 9, 4073 CrossRef; (b) S. C. Kim, D. K. O'Flaherty, L. Zhou, V. S. Lelyveld and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 13318–13323 CrossRef CAS.
- (a) C. Hättig, Advances in Quantum Chemistry, 2005, vol. 50, pp. 37–60 Search PubMed; (b) TURBOMOLE V7.3 2018, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com Search PubMed.
- O. Kennard, J. Biomol. Struct. Dyn., 1985, 3, 205–226 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, computed vertical excitation spectra, excited-state PES calculated for the HC base pair, description of MD simulations and Cartesian coordinates of key stationary points. See DOI: 10.1039/c9cc06180k |
| This journal is © The Royal Society of Chemistry 2020 |