
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA folds threaten genetic stability and can be leveraged for chemotherapy
Joanna
Zell
a,
Francesco
Rota Sperti
a,
Sébastien
Britton
 *bc and
David
Monchaud
*a
*bc and
David
Monchaud
*a
aInstitut de Chimie Moléculaire de l’Université de Bourgogne, ICMUB CNRS UMR 6302, UBFC Dijon, France. E-mail: david.monchaud@cnrs.fr
bInstitut de Pharmacologie et de Biologie Structurale, IPBS, Université de Toulouse, CNRS, UPS, Toulouse, France
cÉquipe Labellisée la Ligue Contre le Cancer 2018, Toulouse, France. E-mail: Sebastien.Britton@ipbs.fr
First published on 30th September 2020
Abstract
Damaging DNA is a current and efficient strategy to fight against cancer cell proliferation. Numerous mechanisms exist to counteract DNA damage, collectively referred to as the DNA damage response (DDR) and which are commonly dysregulated in cancer cells. Precise knowledge of these mechanisms is necessary to optimise chemotherapeutic DNA targeting. New research on DDR has uncovered a series of promising therapeutic targets, proteins and nucleic acids, with application notably via an approach referred to as combination therapy or combinatorial synthetic lethality. In this review, we summarise the cornerstone discoveries which gave way to the DNA being considered as an anticancer target, and the manipulation of DDR pathways as a valuable anticancer strategy. We describe in detail the DDR signalling and repair pathways activated in response to DNA damage. We then summarise the current understanding of non-B DNA folds, such as G-quadruplexes and DNA junctions, when they are formed and why they can offer a more specific therapeutic target compared to that of canonical B-DNA. Finally, we merge these subjects to depict the new and highly promising chemotherapeutic strategy which combines enhanced-specificity DNA damaging and DDR targeting agents. This review thus highlights how chemical biology has given rise to significant scientific advances thanks to resolutely multidisciplinary research efforts combining molecular and cell biology, chemistry and biophysics. We aim to provide the non-specialist reader a gateway into this exciting field and the specialist reader with a new perspective on the latest results achieved and strategies devised.
 Joanna Zell | Dr Joanna Zell obtained her MChem degree from the University of Sheffield (UK) supervised by Dr David M. Williams. She moved to Paris (France) for her PhD at the Institut Curie under the supervision of Drs Ludger Johannes and Frederic Schmidt, developing copper-free clickable tools to reconstitute glycolipids into live cells, defended in 2018. She joined the ICMUB Dijon (France) in 2019 to study the therapeutic relevance of three-way DNA junctions as anticancer targets. In parallel she is an active member of the Chimistes sans Frontières and the Franco-British Council Local Leaders. |
 Francesco Rota Sperti | Francesco Rota Sperti obtained his BSc degree in chemistry from the Universita di Pavia (Italia). He then moved to Orsay (France) to complete his MSc degree in the group of Dr Daniela Verga at the Institut Curie, dealing with the use of c-exNDI derivatives as G-quadruplex sensors. He joined the ICMUB Dijon in Oct. 2019 to complete his PhD on quadruplexes, being in charge of the design, the synthesis and the study of ever smarter template-assembled synthetic G-quartet (TASQ)-based biomimetic G-quadruplex ligands. |
 Sébastien Britton | Dr Sébastien Britton received his PhD in 2009 (University of Toulouse, France) on the study of DNA repair. Then he joined Prof. Steve Jackson's laboratory (Cambridge, UK) where he was initiated to chemical biology. In 2013, he moved to Dr Patrick Calsou's lab (Toulouse, France) to characterise the interplay between DNA repair mechanisms, and the mechanism of action of small molecules. In 2015, he was appointed as a CNRS researcher and in 2018 he was awarded the CNRS Bronze Medal for his discoveries. Starting in 2021, he will lead the “Deciphering and Drugging DNA Repair” team at IPBS (Toulouse, France). |
 David Monchaud | Dr David Monchaud received his PhD in chemistry 2002 at the University of Geneva (Switzerland) under the supervision of Prof. Jérôme Lacour. After two post-docs in Paris (France), he was appointed as a CNRS researcher in 2005 in the laboratory of Prof. Jean-Marie Lehn (College de France, Paris) under the supervision of Dr Marie-Paule Teulade-Fichou, before moving to Institut Curie (Orsay) in 2007 with Dr Teulade-Fichou, and to the Institut de Chimie Moléculaire de l’Université de Bourgogne (ICMUB, Dijon, France) in 2009, to develop chemical genetics programs on DNA/RNA secondary structures, smart ligands and probes. |
Introduction
High levels of genetic instability and mutations is a general enabling hallmark of cancer, as defined by Douglas Hanahan and Robert A. Weinberg in their cornerstone reviews.1,2 This is likely a result of the need for tumour progression to acquire multiple mutations, e.g. in oncogenes and tumour-suppressor genes, for successful tumour initiation, and potentially metastasis. The genome maintenance systems limit such genetic defects in healthy cells but this is often impaired in cancer cells, making the accumulation of DNA lesions a common feature in cancer. Cancer cells’ reduced ability to repair DNA damage, driving their genomic instability, provides an important vulnerability that is exploited therapeutically.3–5Recent advances in genetics, genomics and proteomics have provided a better understanding of the complex interplay between DNA lesions and repair mechanisms, and what is therapeutically relevant to halt cancer cell proliferation.6 These advances have generated momentum for designing more selective and efficient anticancer strategies, often relying on combinations of DNA damaging agents and DNA repair inhibitors.5,7,8 Recent advances in our understanding of the complex aspects of DNA/RNA secondary structure9–12 provide brand new opportunities to exploit DNA damage in a more precise manner. Alternative nucleic acid structures are defined as structures that deviate from the canonical Watson–Crick DNA double helix, B-DNA or duplex-DNA. From a molecular viewpoint, the selective small-molecule targeting of such DNA structures holds potential over gene sequence targeting, given that they present well-defined 3D-structures,13,14 in a manner reminiscent of modern pharmaceutical targeting of proteins and enzymes.15 Furthermore, alternative DNA structure formation is coupled with DNA transactions (replication, transcription) due to transient strand separation and local DNA deformation.16–18 Such structures thus offer the added advantage of creating replication/transcription-associated DNA damage whose repercussions will be felt primarily in highly proliferating cells. The present review aims at gathering the most recent results obtained in this new and promising approach, to demonstrate the relevance of targeting higher-order DNA structures to treat cancer.
1. The discovery of DNA as a chemotherapeutic target
DNA is constantly exposed to damaging agents – “tens of thousands of DNA lesions per day”3 – of both endogenous and exogenous origins.32,33 Beyond irradiation and cross-linking chemicals, different types of lesion have now been unravelled, which include chemical modifications (e.g. oxidations, deaminations), DNA replication errors (e.g. mismatched Watson–Crick base pairs) and DNA transaction impediments (e.g. non-B helix structured DNA, sometimes called ‘difficult-to-replicate’ sequences34,35) which can stall replication and transcription (Fig. 1). The plurality of DNA damage explains why cells have evolved such a highly sophisticated and efficient network of surveillance, signalling and repair pathways, collectively referred to as the DNA damage response (DDR).3,4,33,36,37 The molecular complexity of the DDR is just starting to be understood, because of the multiplicity of pathways involved, including excision (BER, NER, MMR), recombination (HR) and joining (NHEJ) pathways (see Sections 2a and b) and their constant and intricate cross-talk. DDR ultimately aims at controlling genomic instability in normal cells, whilst from a therapeutic viewpoint, the DDR presents a key strategic Achille's heel of cancer cells, which can be targeted to impede their anarchic proliferation.
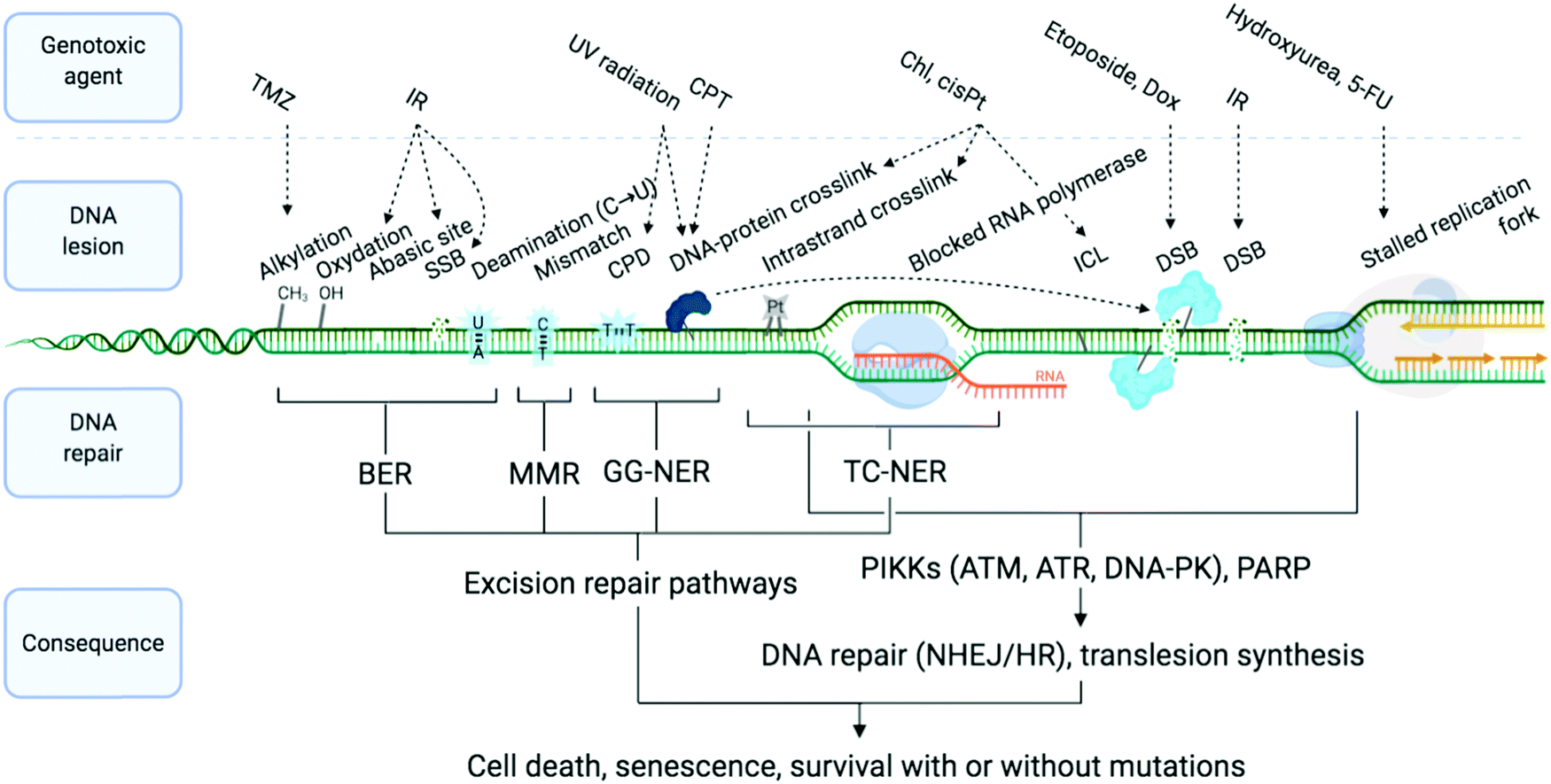 | ||
| Fig. 1 Examples of DNA-damaging agents, the lesion which they induce, and the downstream consequence. Temozolomide (TMZ), ionising radiation (IR), camptothecin (CPT), chlorambucil (Chl), cisplatin (cisPt), doxorubicin (Dox), single-strand break (SSB), double strand break (DSB), cyclobutane pyrimidine dimer (CPD), interstrand cross-link (ICL), base excision repair (BER), mismatch repair (MMR) and global genome nucleotide excision repair (GG-NER), transcription-coupled NER (TC-NER). Adapted from ref. 70, created with BioRender. | ||
These results gave new impetus to the quest for small molecule therapy. Similarly to Gilman and Goodman, Sidney Farber made the link between the high lymphocyte levels observed in leukaemia patients’ blood and potentially leukopenic vitamins antifolates, who began pioneering trials on last-chance child leukaemia patients with synthetic antifolates in 1947.48,50,51 These research efforts led to the first example of targeted therapy with the use of 5-fluorouracil (5-FU) in 1957,52 which specifically targets the abnormal uracil dependence of hepatomas.53 The benchmark example of targeted therapy, imatinib (Gleevec) was reported in 1996,54 being the first approved anticancer drug molecularly designed for its specific protein target, the constitutively active BCR-ABL tyrosine kinase produced by the Philadelphia chromosome in chronic myelogenous leukaemia.55
The most defining characteristic of cancer cells is their uncontrolled growth, and this is exploited by antiproliferative agents. This class of chemicals encompasses the oldest and still most popular strategy of chemotherapy. Such agents now include several major subgroups (Fig. 1): chemicals which directly alkylate the DNA such as bifunctional cross-linking agents (nitrogen mustards including chlorambucil (Chl) and platinum drugs including cisplatin (cisPt), carboplatin and oxaliplatin) which cause intrastrand cross-links (and less frequently interstrand and DNA–protein cross-links)56,57 and monofunctional alkylators such as temozolomide (TMZ); topoisomerase poisons such as etoposide, which intercalates at the breakage site formed as DNA topoisomerase 2 (TOP2) induces a DSB to disentangle DNA, thus trapping the DNA–TOP2 cleavage complex,58–60 and camptothecin (CPT) and derivatives (topotecan, irinotecan), which similarly trap the DNA–topoisomerase 1 (TOP1) complex;61,62 antimetabolites (inhibitors of nucleotide metabolism such as folate antagonist methotrexate, and 5-FU);63 and antimitotic agents such paclitaxel that impedes progression through mitosis by targeting tubulin.
It is widely accepted that cytotoxic agents can attack normal cells as well. In order to treat patients whilst reducing extreme side-effects, drug combinations are often used, accompanied by surgery and radiotherapy of solid tumours. The first combination of DNA targeting agents associated several cytotoxic drugs (e.g. etoposide and cisPt for small-cell lung carcinoma or FOLFIRINOX protocol for metastatic pancreatic cancers).
Another hallmark of cancer cells is their genetic instability, notably caused by an imbalance in DDR mechanisms.2,64 Among the most documented examples of oncogenic dysregulations, BRCA1/2 and TP53 mutations rank highly. Mutations in Breast cancer susceptibility genes BRCA1 and BRCA2 are associated with breast and ovarian cancers.65,66 BRCA1/2 play a critical role in DSB repair by homologous recombination (HR), a key DDR pathway (see Section 2a2). TP53, the gene encoding the apoptosis- and DNA-damage-checkpoint-regulating protein p53, is the most frequently mutated gene in cancer, at approximately half of all cancers.67,68 It is classically proposed that these genetic dysregulations are advantageous to tumour progression,2 and in a recent study, most cancers analysed from The Cancer Genome Atlas were enriched in mutations coding for DDR proteins.69 For this reason, modern therapeutic strategies that target DDR, and combinations of DNA-damaging agents and DDR inhibitors, are evolving rapidly (discussed in Section 2d).
2. The DNA damage response (DDR)
The DNA damage response (DDR) is a broad term that designates the mechanisms involved in the response to all DNA lesions. However it is frequently used in a restrictive manner to designate the mechanisms responding to DNA double-strand breaks (DSB). Indeed, owing to their high cytotoxicity, DSBs induce a robust response that is initiated by the detection of the lesion by PIKKs and/or PARPs (see Section 2b) and that leads to (1) activation of cell cycle checkpoints that blocks or slow down cell cycle progression at specific boundaries; (2) DSB repair that is principally mediated by HR and NHEJ, and (3) activation of a gene expression program that dictates long term response. The DDR can result in cell survival when the amount and type of DSBs is manageable, or in senescence or cell death when damage is too severe (Fig. 2).3,71,72 DNA repair mechanisms can be faithful or instead fix mutations in the genome. While DSB-induced genetic variations can be deleterious, with for example the formation of translocations that can be responsible for secondary cancers, they can also be a desired outcome of several physiological processes. This is the case for antibody diversification by variable (diversity) joining (V(D)J) recombination and class-switch recombination (CSR) in which recombination-activating gene (RAG) nuclease for V(D)J, and activation-induced cytidine deaminase (AID) for CSR promote sites-specific DSBs, and for meiotic recombination which relies on SPO11 for genome-wide DSBs formation to promote recombination between homologous chromosomes.73–75 The DDR can be activated by bona fide DSBs (leading to ATM and DNA-PKcs activation), by accumulation of single-stranded DNA (leading to ATR activation), by conversion of a DNA lesion into one these structures as a result of defective DNA repair, or by conversion of stalled DNA replication or transcription complexes.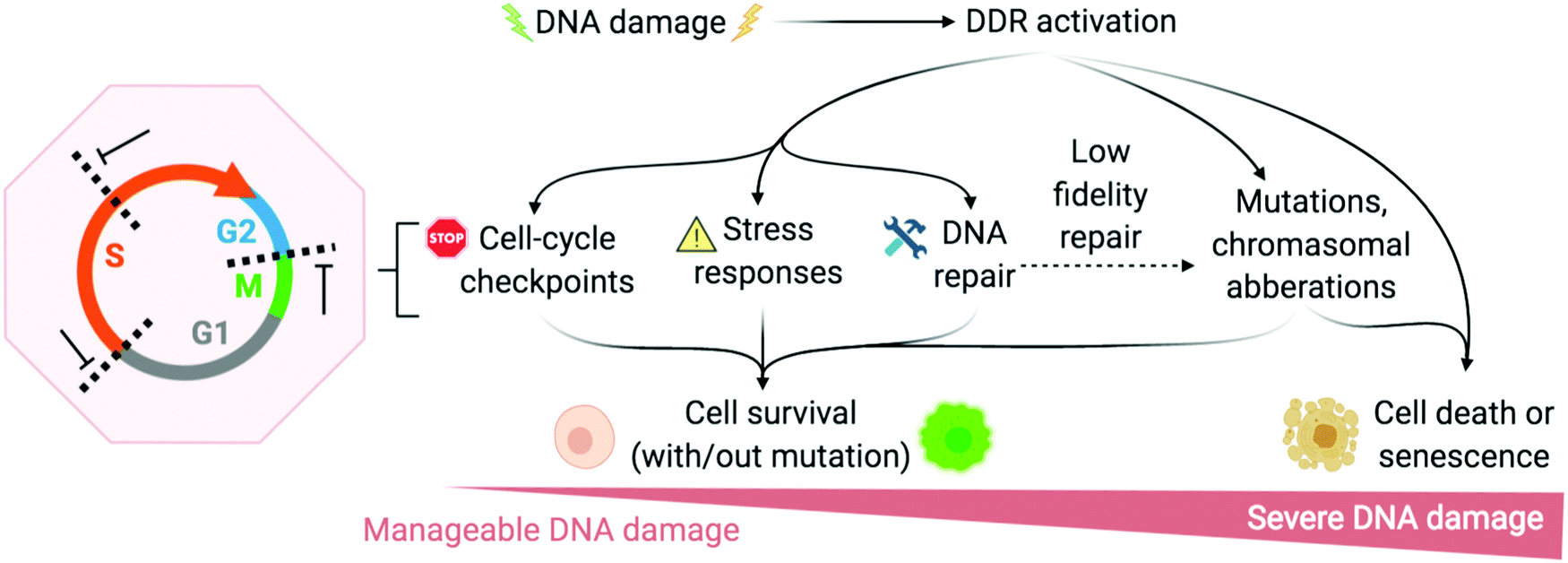 | ||
| Fig. 2 DNA damage response (DDR) is activated to varying degrees depending on the extremity of DNA damage. Adapted from ref. 72, created with BioRender. | ||
2a1. Single-strand breaks and damaged/mismatched nucleotides repair. Tomas Lindahl, Paul L. Modrich and Aziz Sancar were awarded the Nobel prize in chemistry in 201576–78 for their pioneering work describing DNA repair mechanisms, including base excision repair (BER), mismatch repair (MMR) and nucleotide excision repair (NER). Multiple repair mechanisms exist for all types of DNA damage (Fig. 1),3,33,79,80 and a large degree of overlap exists within the multiple DNA repair pathways and with double-strand break (DSB) repair pathways.69 Single-strand breaks (SSBs) are the most common DNA damage occurring throughout a cell's lifetime, with a large amount being generated endogenously by reactive oxygen species (ROS) generating SSB directly or resulting in DNA lesions processed by DNA glycosylases and apurinic/apyrimidinic endonucleases (APEs). We provide bellow a brief description of the SSB repair pathways:
BER: small nucleobase lesions such as alkylations, oxidations and deaminations are repaired by BER, in which a single nucleobase is removed in a concerted dual excision by a DNA glycosylase (e.g. 8-oxoguanine DNA glycosylase 1, OGG1) that specifically recognises the base adduct, and an endonuclease recruited to the abasic site (e.g. APE1). The SSB thereby generated is recognised by the crucial mediator protein PARP-1 that promotes the assembly of several proteins, including SSB repair enzymes, through the formation of poly-ADP ribose chains on local DNA-associated proteins (see Section 2b). Depending on the DNA lesion, short- or long-patch BER will process the damage. In short-patch BER, in which a single nucleotide is repaired, the abasic site is filled by DNA polymerase β and sealed by DNA ligase 3 (Lig3) in complex with the X-ray repair cross-complementing protein 1 (XRCC1).32 In long-patch BER, where 2–12 nucleotides must be replaced due to a bulkier DNA lesion, DNA polymerases (DNA Polδ, ε or β) act to fill the gap in concert with the action of flap endonuclease 1 (FEN1) that processes the resulting flap intermediate and DNA ligase I (Lig1) that seals the nick.80
MMR: the mismatch recognition protein complex Mutator S alpha (MutSα), heterodimer of mutS homologues 2 and 6 (MSH2/MSH6), senses Watson–Crick base mismatch protrusions formed from imperfect DNA replication. It moves with DNA replication machinery including the proliferating cell nuclear antigen (PCNA) and DNA Polδ, although it may or may not be coupled directly, scanning the newly synthesised DNA for mismatches in leading and lagging strands.81,82 At a point of damage, the endonuclease complex MutLα, heterodimer of MutL homologue 1 (MLH1) and the post-meiotic segregation increased 2 (PMS2) proteins are recruited and forms a nick on either the 3′ or the 5′ side of the damage. From this nick, exonucleases, primarily Exo1, remove a stretch of 1 to 2 kilobases (kb) from the damaged strand. The exposed single-stranded DNA (ssDNA) is stabilised by Replication Protein A (RPA) and the gap is filled by DNA replication machinery (PCNA-DNA Polδ) and ligated by Lig1. New mediators and damage substrates of this pathway are still being discovered, and this pathway partly overlaps with NER, for which reason Sancar once warned against the rigid, over-simplified one-damage-one-pathway classification.83
NER: bulkier, helix-distorting lesions such as CPD, intrastrand cross-links (covalent linkages between two bases of the same strand) and small DNA–protein cross-links (<8–10 kDa)61,84,85 are repaired by NER, which is further categorised into transcription-coupled (TC-NER) and global genome (GG-NER) repair pathways, differing in when and where in the cell cycle they occur and in the initial sensor proteins they involve. TC-NER occurs in rhythm with the advancing RNA polymerase (RNAP) and is initiated when the transcription complex is blocked by other lesions.86 Unlike TC-NER, GG-NER operates throughout the genome after recognition by the protein sensors xeroderma pigmentosum group C (XPC) or group E (XPE) coupled with the damage DNA binding protein 2 (DDB2). These distinct sensors recruit the same downstream NER mediators: first, a ternary complex that includes the transcription factor IIH (TFIIH) and two helicases, xeroderma pigmentosum group B and group D (XPB and XPD) that unwind the damaged DNA and create a ∼30 nucleotide bubble; then, an endonuclease complex comprising the xeroderma pigmentosum group F (XPF) and excision repair cross-complementation group 1 (ERCC1) proteins incises on the 5′ end of the bubble while the xeroderma pigmentosum group G (XPG) endonuclease incises at the 3′ end, releasing an oligonucleotide carrying the DNA lesion. Like in MMR, the gap is then filled by DNA Polε and Polδ, the leading and lagging strand polymerases respectively87 and ligated (Lig1).78,80,88 GG-NER is not constitutive like TC-NER, but is induced during DNA-damage repair signalling, and is less sensitive to certain DNA adducts than TC-NER.70
Direct repair: certain common methylation adducts are reversed in a single step, such as methyl group transfer (O6-methylguanine) or oxidative demethylation (1-methyladenine and 3-methylcytosine), by O6-meG-DNA methyltransferase (MGMT) and 1meA/3meC-DNA dioxygenase respectively (AlkB homologues).89
2a2. Double-strand breaks (DSBs) repair. As compared to SSBs, DSBs pose a more serious threat to genetic integrity since they result in the loss of chromosomal integrity which can lead to the loss of chromosome arms, translocations and cell death. DSBs are highly toxic and a single DSB can result in cell cycle arrest90 and cell death.3 DSBs can be generated directly, such as those generated by TOP2 poisoning, ionising radiation, or as a result of the conversion of a primary lesion or blocking structure by transcription or DNA replication.91,92 DSBs are repaired by two principal mechanisms (Fig. 3): the non-homologous end joining (NHEJ) pathway, which joins two DNA ends without a DNA template; and the homologous recombination (HR), in which nucleases strip away a stretch of nucleotides at broken ends, exposing a 3′ overhang of ssDNA in a process called DNA end resection, then use the corresponding sister chromatid as a template to repair the damaged strand.6,37,93
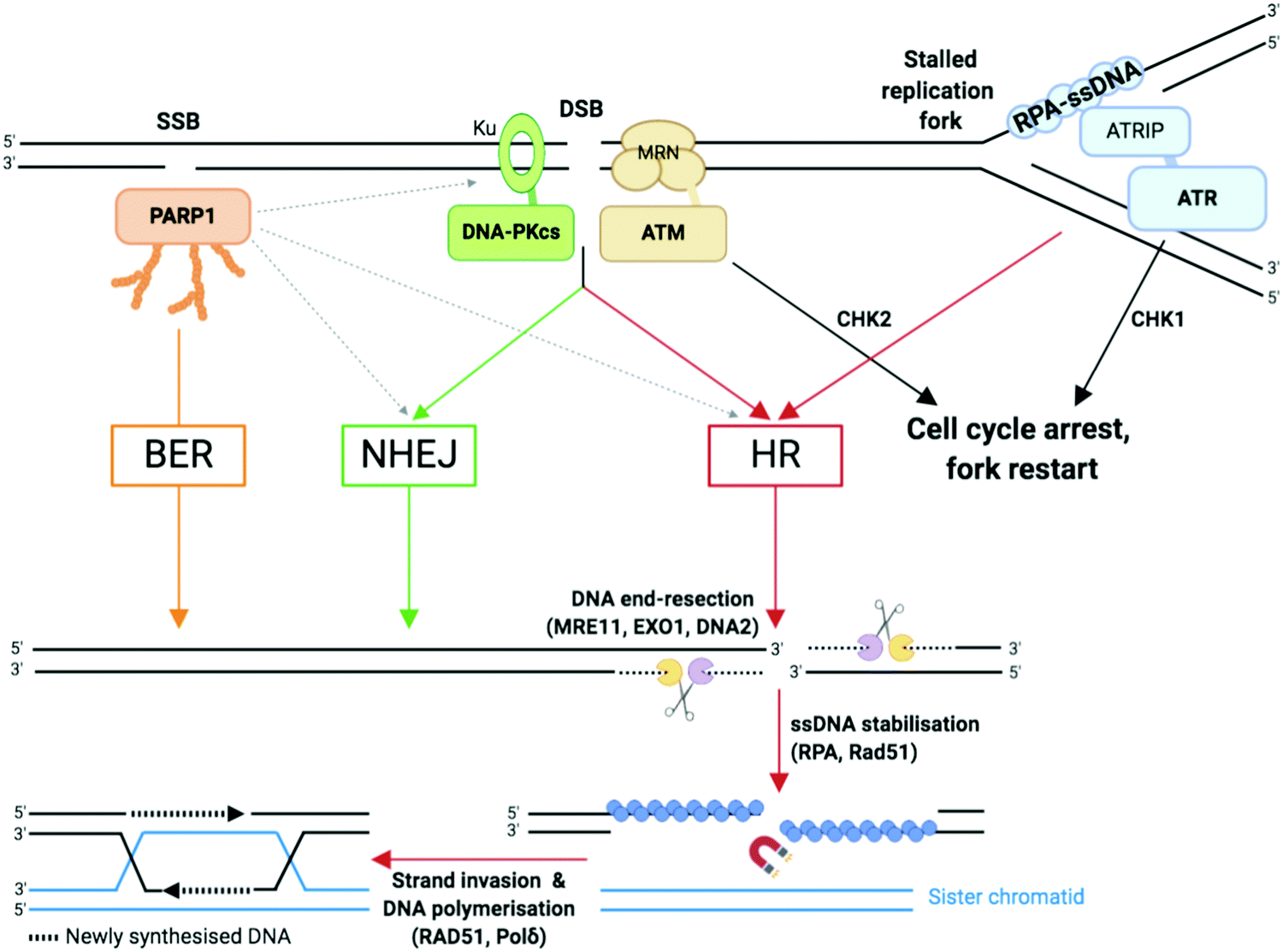 | ||
| Fig. 3 Single-strand breaks (SSB) and double-strand breaks (DSB) repair pathways. Dotted arrows indicate that PARP1 can be activated by DSB and stimulate DSB repair by both HR and NHEJ.33,109 Adapted from ref. 37, created with BioRender. | ||
NHEJ: NHEJ is initiated when the DNA ends are recognised by the Ku complex, the heterodimer of Ku70 and Ku80.33 Ku shields the DNA ends from exonuclease activities and is a hub for recruiting the other NHEJ factors, including the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs). Multiple DNA processing enzymes are involved in cleaning the DNA ends for repair by NHEJ, including Artemis (DNA cross-link repair 1C, DCLRE1C), the X polymerases Polμ and Polλ, and the polynucleotide kinase/phosphatase (PNKP). Finally, the DNA ends are sealed by the action of the ligation complex composed of XLF–XRCC4–DNA ligase 4 (Lig4). Since NHEJ does not rely on nucleotide sequence matching and it allows short deletions or insertions upon repair, it is considered an error-prone repair mechanism. And yet, sequence fidelity is mostly maintained with little loss in genetic material or chromatin rearrangement, likely thanks to fast recognition of blunt ends of DSB by highly abundant sensor proteins such as Ku, which keeps breaks in close proximity to be repaired.94 NHEJ occurs throughout the cell cycle and is the most prevalent DSB repair mechanism in non-dividing cells.
HR: HR is initiated by the generation of a 3′ ended ssDNA extension produced by exonucleolytic processing of the DNA ends. This process, called DNA end resection, is controlled by the nuclease activity of MRE11 (Fig. 3). MRE11, as part of the MRE11–RAD50–NBS1 (MRN complex) associates to the chromatin flanking each DSB, through ATP-dependent RAD50 DNA clamping activity. CtIP (C-terminal binding protein 1 (CtBP)-interacting protein, also known as retinoblastoma-binding protein 8, RBBP8), an interaction partner of MRN, controls MRE11 endonuclease activity which is stimulated when CtIP is phosphorylated by CDKs and ATM/ATR (see Section 2b).95 This triggers the formation of a nick which is the initiation site for bidirectional resection performed in the 3′–5′ direction by the exonuclease activity of MRE11 and in the 5′–3′ direction by the exonuclease activity of EXO1 and/or DNA2 in complex with the Bloom syndrome helicase (BLM). Following end resection, the ssDNA is stabilised by RPA, which comprises the heterotrimer RPA1/2/3 known to rapidly bind to and stabilise ssDNA via its oligonucleotide/oligosaccharide-binding fold (OB-fold) domains during DNA replication and repair96 (Fig. 3). RPA is replaced by Rad51 (homologue of bacterial recombination protein A, recA), responsible for scanning the sister chromatid for the homologous strand, aided by BRCA2 and BRCA1 in association with BRCA1-associated RING domain 1 (BARD1).97,98 Homologous strand invasion requires stabilisation of the non-template sister strand by Rad51, whilst DNA Polδ fills in the invading strand using the template strand (Fig. 3). HR occurs during and after synthesis (S) phase, when two sister chromatids are present, and enables faithful DNA replication (Section 2b).
2a3. Interstrand cross-links (ICLs) repair. ICLs are covalent linkages between two different DNA strands, as opposed to intrastrand cross-links that occur on the same strand and which are primarily repaired by NER. ICLs are detected and repaired during DNA replication by the Fanconi Anemia (FA) pathway, named after the disease resulting from genetic mutations of key components of this pathway. The FA pathway requires activation by ATR kinase, a master mediator of DDR signal transduction (see Section 2b), which simultaneously reduces replication fork speed.99–101 Currently, 22 genes have been described as FA genes, some of them being implicated in other DNA repair mechanisms: FANC-A, -B, -C, -D1 (BRCA2), -D2, -E, -F, -G (XRCC9), -I, -J (BRIP1), -L (PHF9), -M, -N (PALB2), -O (RAD51C), -P (SLX4), -Q (ERCC4/XPF), -R (RAD51), -S (BRCA1), -T (UBE2T), -U (XRCC2), -V (REV7/MAD2L2) and -W (RFWD3).102 The FANC-A, -D, -C, -E, -F, -G, -L and -M proteins form the core complex which monoubiquitinates FANCD2/FANCI upon ICL recognition, while the other proteins mediate the lesion incision or “unhooking” forming a DSB whose repair involves components of translesion synthesis (TLS, DNA polymerase κ) and HR pathways. The FA complex recognises the ICL, often at the collision site of one or two stalled replication forks, leading to incision by endonucleases (the structure-specific endonucleases SLX1, MUS81 (also known as SLX3), XPF and ERCC1) in one strand on either side of the ICL thus “unhooking” the ICL lesion and forming a DSB. Replicative polymerases act on the intact strand to fill the template whilst retaining the incised strand in proximity. Finally the HR machinery repairs the DSB in the incised strand from the template double strand.103 ICLs can also be repaired by a replication-independent mechanism involving incisions on the ICL flanks by the NER machinery and lesion bypass by Polκ.99,104,105
PARP1 and PARP2 (the latter accounting for 5–10% of total PARP activity)106 are considered a major first line of defence in the DDR response.4,33,37 PARP activation, PARylation (synthesis of branched and linear chains of poly(ADP-ribose) (PAR) on proteins) and auto-PARylation (Fig. 4a) occurs rapidly at sites of SSB, DSB and stalled replication forks.80,107 PAR attachments on PARP and on nearby histones act as a platform promoting the recruitment of DDR factors including XRCC1/Lig3 and inducing chromatin remodelling.108 Importantly, PARP1 is also activated by DSBs where it promotes the recruitment of NHEJ and HR factors.33,109 Repair of SSB can either be processed in a short- or long-patch DNA gap filling mechanism similar to that of BER,107 mediated by nucleases such as FEN1 and DNA Polδ, the lagging strand polymerase capable of strand displacement.87 The role of PARP3 in DNA repair is more poorly understood, although has been shown to have more protein PARylation targets than PARP1 and 2 with minor overlap.110 PARP3 has a different DNA-binding domain, it is described to recognise DSB only111 and it has been shown to promote the recruitment of the NHEJ ligation machinery, the XRCC4/Lig4 complex.112 Of interest, PARP3 was found to promote chromosome rearrangements and its genetic knockout in lung adenocarcinoma cells showed extreme sensitivity to small molecules which induce DSB113 or stabilise the secondary DNA structure G-quadruplexes (G4).114
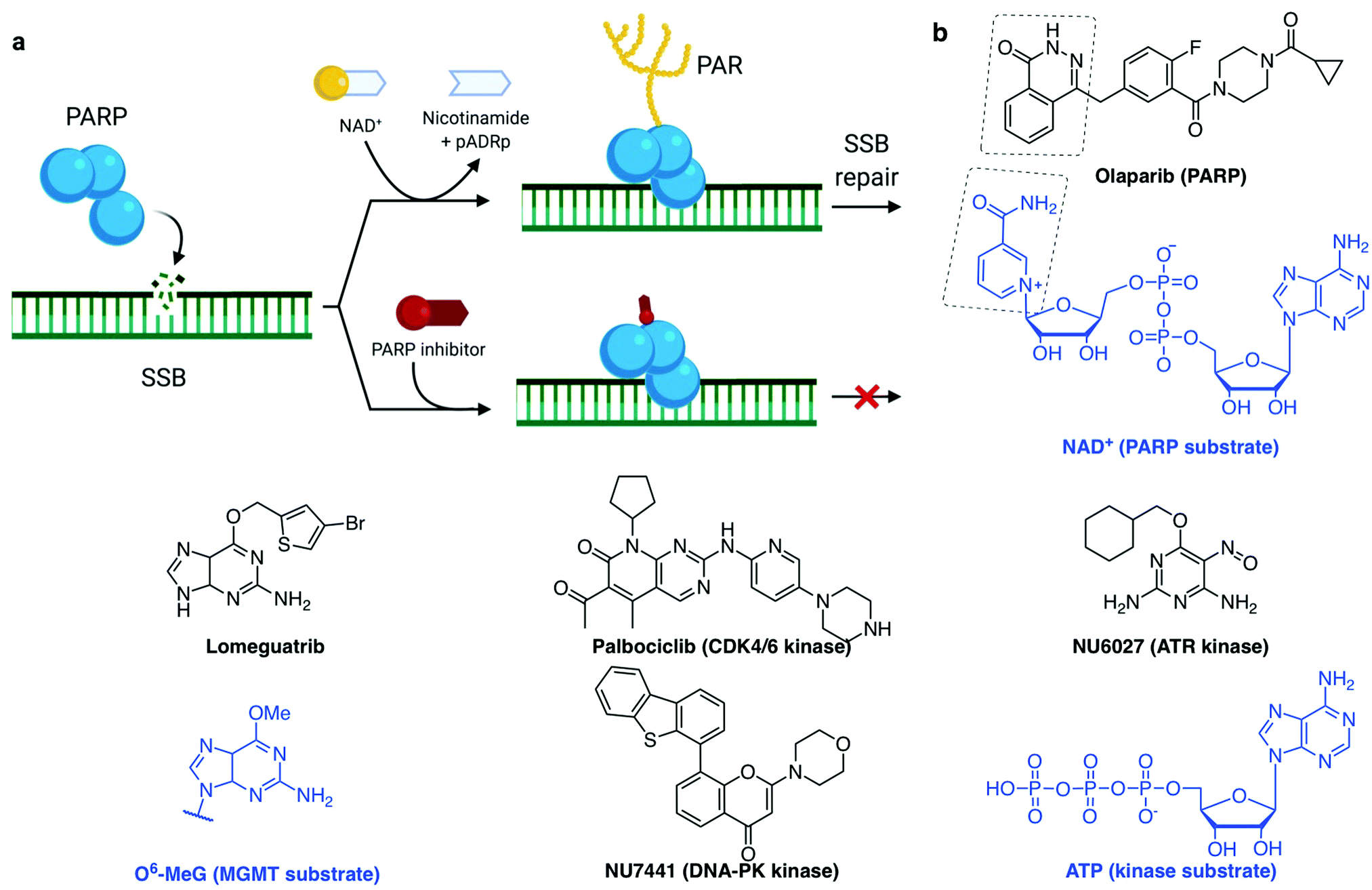 | ||
| Fig. 4 (a) PARP recruitment to SSB and synthesis of poly(ADP-ribose) (PAR) to activate SSB repair. Inhibition of repair by PARP-trapping at the break site. (b) Examples of inhibitors (with their protein target) and the substrate they mimic (blue). (a) Created with BioRender. | ||
The PIKKs coordinate the central alarm system to DSB and stalled forks, often during NHEJ/HR (see Section 2a2), through activation by (auto-)phosphorylation and recruitment of a vast range of signalling molecules.33,115 These signal transducers have significant structure and function similarity, however are activated by distinct sensor proteins which recognise the DNA damage. PIKKs comprise two families of effectors, DNA dependent protein kinase (DNA-PK) on one hand, and ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad3-related (ATR) on the other hand, which are recruited to DNA damage sites through molecular interactions with analogous C-terminal motifs of Ku80, Nibrin (or Nijmegen) breakage syndrome gene (NBS1) and ATR-interacting protein (ATRIP) respectively (Fig. 3):37,116
– DNA-PKcs (also known as DNA dependent protein kinase catalytic subunit) is involved in DSB repair by NHEJ. The Ku heterodimer comprising Ku70/Ku80 (also known as X-ray repair cross-complementing protein 6 and 5, XRCC6 and XRCC5, respectively) binds to free DSB ends (amongst other DNA structures) and recruits DNA-PKcs. The Ku–DNA-PKcs–DNA complex forms the active DNA-PK holoenzyme which is a serine/threonine kinase able to phosphorylate several substrates including itself. Autophosphorylation of DNA-PKcs regulates DNA end processing.
– ATM, through a specific amplification loop, plays an apical role in DSB signalling and cell cycle checkpoint activation, in addition to its function in promoting DSB repair via HR.6,71 The MRN complex associates to the flank of each DSB through the ATP-dependent RAD50 clamp which recognises the DNA damage. The MRN complex mediates the recruitment of ATM by direct interaction between ATM and the NBS1 C-terminus.117 However, in contrast to Ku whose recruitment is limited to one or two Ku proteins per DSB end,118 multiple MRN–ATM complexes are recruited to chromatin thanks to an amplification loop relying on ATM kinase activity. Indeed, ATM is responsible for the phosphorylation of a large number of proteins including the C-terminus of the histone variant H2AX, generating γH2AX, the S139-phosphorylated form of H2AX. This initial phosphorylation recruits MDC1 (mediator of DNA damage checkpoint protein 1), via its BRCA1 C terminus (BRCT) repeat domains,119–122 which is itself phosphorylated by casein kinase 2 (CK2). The phosphorylated form of MDC1 is recognised by the forkhead-associated (FHA) and BRCT domains of NBS1 which drives the secondary recruitment of the MRN–ATM complex that subsequently phosphorylates other more distant substrates including H2AX.123–126 This amplification loop leads the γH2AX signal to spread from kilo- to megabases from the DSB, making the detection of the γH2AX signal a sensitive and widespread approach to monitor the induction of DNA damage in cells (examples in Section 3c). The rapid spreading of γH2AX from DSB sites is controlled by chromatin domain boundaries delimited by DNA-binding cohesin protein complexes,127,128 which also repress transcription of damaged genes.129 In addition to NBS1, MDC1 recruits other proteins that govern cell cycle checkpoints, such as checkpoint kinase 2 (CHK2), and mechanisms of DNA repair pathway choice including 53BP1 (promoting NHEJ) and BRCA1 (promoting HR).130,131 As well as recruiting DNA repair proteins (MRN/ATM), γH2AX recruits chromatin remodelling machinery to aid damage repair.122,132 It is noteworthy that ATM can also be directly activated by oxidative stress via oxidation of some of its cysteines.71 In parallel to its signaling functions, ATM also stimulates HR repair (Fig. 3) by phosphorylating several proteins including the MRN interacting protein CtIP. CtIP phosphorylations promotes MRE11 endonuclease activity, responsible for nicking the side(s) of the DSB to initiate end resection. The ATM-dependent CtIP phosphorylations promote Ku eviction from the DSB end, thereby directing DSB repair towards HR.133 In addition, ATM stimulates HR by phosphorylating several HR factors, such as BRCA2, EXO1 and BLM, and NHEJ factors such as XRCC4, XRCC4-like factor (XLF) and DNA-PKcs.134–137 ATM also plays a crucial role in enforcing the cell cycle checkpoints through activation by phosphorylation of CHK2, which in turn activates p53, a key apoptosis regulator.68 53BP1 is recruited to nucleosomes with specific histone modifications: Ring Finger Protein 168 (RNF168)-ubiquitylated H2AK15 and dimethylated H4K20;138 where it functions as a central determinant in the repair pathway choice made at DSB by promoting the recruitment of the shieldin complex,139 thereby limiting DNA end resection by BRCA1/BARD1, which exposes ssDNA to be repaired by HR. As such 53BP1 inhibits HR and supports NHEJ repair in the G1-phase of the cell cycle.6,140 PARP3, in interaction with Ku, similarly is described to favour DSB repair to NHEJ by limiting end resection.113
– ATR is the key damage signalling mediator at replication forks and in dividing cells. ATR binds through ATRIP to the RPA:ssDNA complex,141,142 which forms rapidly at DNA replication fork blockages during the S phase of the cell cycle (Fig. 2 and 3) and stabilises ssDNA during DNA replication. Upon DNA damage, ATR induces a signalling cascade, phosphorylating a long list of cell cycle-related substrates, notably checkpoint kinase 1 (CHK1) and apoptosis and replication stress regulator p53.4,143 ATR activation stabilises the stalled fork (preventing fork collapse which can otherwise lead to DSB formation)144 and prevents proximal replication origin restart.142,145,146 Stalled replication forks can be restarted by an HR-dependent mechanism. Unlike ATM and DNA-PKcs, ATR is necessary for cell growth and embryonic development,147,148 meaning mechanistic studies of ATR were greatly facilitated by the recent development of ATR-inhibiting chemicals. Cross-talk exists between the ATM and ATR kinases since ATR can activate ATM in some instances,149 the two kinases having many substrates in common and their cellular roles strongly overlapping. However, most of the details regarding the intricate genetic regulations in which ATM and/or ATR remain to be discovered.
Synthetic lethality describes the relationship between two gene deactivations which cause cell lethality when both genes are silenced (through endogenous mutation, exogenous inhibition or knockdown) but cells are viable when only one is deactivated.7,162–165 As discussed above, DDR pathways function with significant redundancy, yet with one pathway missing, cancer cells become more dependent on the remaining pathways. A myriad of clinical trials are in place to assess the benefit of inhibiting multiple DDR pathways concomitantly, with the hope of being able to widen the therapeutic window by increasing the combined tolerated dose and decreasing the effective dose.8 The first-in-class example of a synthetic lethality strategy applied to cancer is the PARP inhibitor olaparib (Fig. 4b),8,108 which was approved for treatment of several BRCA-deficient cancer types by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) in 2014. More recently (in 2018), another PARP inhibitor talazoparib was approved as it displays a higher cytotoxicity in BRCA-deficient tumours, likely due to its stronger PARP–DNA trapping affinities (Fig. 4a).166 PARP inhibitors have proved efficacious in multiple cancers with deficient DDR pathways, as well as in maintenance therapy for cancers which have previously responded to antiproliferative agents.5,160 However, various resistance mechanisms to PARP inhibitors have been described, including via the restoration of HR, for example via inactivation of 53BP1, or by increased replication fork stability, for example via inactivation of the junction endonuclease MUS81 or the N-methyltransferase enhancer of zeste homolog 2 (EZH2).109,166,167
Other DDR inhibitors (see Section 2c) have shown potential in the clinic, including inhibitors of specific DNA repair pathways such as DNA-PK inhibitors (M3814 and CC-115, currently in phase I trials168,169) which block NHEJ-dependent DSB repair, and MGMT inhibitors (lomeguatrib, Fig. 4b) which block the removal of alkylated bases. Also in clinical testing are the inhibitors of cell cycle checkpoint mediators ATR, ATM, WEE1 and CHK1, such as M6620, M3541, AZD1775, LY2606368 respectively, for both single-agent and combination therapies.8,170 Despite promising, the therapeutic indexes of these molecules (toxic dose/therapeutic dose) are often limiting, as demonstrated in the cases of lomeguatrib160 and UCN-01 (first specific CHK1 inhibitor developed from the broad-spectrum kinase inhibitor staurosporine155,171). PARP inhibitors had initially been trialled in combination with DNA damaging agent temozolomide and failed due to excessive toxicity, but following these studies, its synthetic lethality properties in BRCA-deficient cancers were uncovered.160 This exemplifies the advantage of synthetic lethality strategies, in which a single agent can be applied without incurring unacceptable collateral damage, by taking advantage of a known weakness in the patient's cancer. With the aid of modern high-throughput and genome-wide sequencing techniques,172 the body of knowledge on genetic biomarkers is being established for each cancer type, opening up the development of specific small molecules suited to personalised cancer therapy.4,162 Thus, our understanding of the cancer genotypes that are likely to be sensitive to selected treatment is growing.
A common trend in the design of small molecule inhibitors is to structurally mimic the biological substrate of the target protein: O6-benzylguanine and lomeguatrib (O6-(4-bromothenyl)guanine) both mimic O6-methylated guanine (O6-MeG), the DNA base lesion repaired by MGMT (O6-methylguanine–DNA methyltransferase, Fig. 4b); olaparib mimics NAD+, the building block used by PARP for PARylation signalling (Fig. 4a); and palbociclib fits into the ATP-pocket of checkpoint kinase CDK4/6173 (Fig. 4b), whereas other kinase inhibitors can mimic the target protein. Some DDR inhibitors, such as NU6027174 and UCN-01175 (ATR and CHK1 inhibitors respectively, Fig. 4b), were initially designed to target CDKs, and the marginal specificity for DDR checkpoint kinases over another kinase was developed by often lengthy medicinal chemistry methods: trial-and-error synthesis, structure–activity relationship (SAR) studies and enzyme assays, since many active site crystal structures of these targets have not yet been resolved.
In the next chapter, we discuss another approach, which could bypass certain toxic and resistance-incurring pitfalls of DDR, or prove synergistic. Inspired by a strategy at the very foundation of chemotherapy, we look again to triggering the DDR by drugs targeting the DNA itself. Accumulating evidence now supports the existence of non-B DNA structures in cells that fold as a result of cellular activity. The resulting higher-order structures are closer to protein than B DNA in terms of 3D structure and offer more structurally defined binding sites for small molecules (e.g. the accessible G-quartets of G-quadruplexes, the central cavity of DNA junctions, see Sections 3a and 4a) than major/minor groove interaction and/or intercalation in between two successive base pairs of B DNA. This offers the possibility of targeting higher-order nucleic acid structures with a better degree of selectivity, thus re-establishing DNA in its many forms as a promising chemotherapeutic target with unique and structurally defined small molecule binding sites. In the following sections, we will describe the new trend that is emerging based on the stabilisation of non-B DNA structures with specifically designed small molecules (ligands), as a way to damage cancer cells’ DNA in a more specific manner.
3. G-Quadruplexes (G4s) as targets to induce DNA damage
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 by G4-seq.183 These sequences are predominantly maintained in an unfolded state, as exemplified by the detection in live cells of only >10000 G4s by G4 ChIP-seq184 and ca. 3000 G4s by the fluorophore SiR-PyPDS (see Section 3c),185 with a strong correlation between individual G4 formation and the transcriptional activity of the gene they fold from. This transiency originates from the various mechanisms the cell has evolved to regulate G4 formation, among which the G4 helicases are being actively studied (see Section 3b). Interestingly, the QFS distribution is not random as they are significantly enriched in key regulatory regions including gene promoters, replication origins and telomeres.10,186
000 by G4-seq.183 These sequences are predominantly maintained in an unfolded state, as exemplified by the detection in live cells of only >10000 G4s by G4 ChIP-seq184 and ca. 3000 G4s by the fluorophore SiR-PyPDS (see Section 3c),185 with a strong correlation between individual G4 formation and the transcriptional activity of the gene they fold from. This transiency originates from the various mechanisms the cell has evolved to regulate G4 formation, among which the G4 helicases are being actively studied (see Section 3b). Interestingly, the QFS distribution is not random as they are significantly enriched in key regulatory regions including gene promoters, replication origins and telomeres.10,186
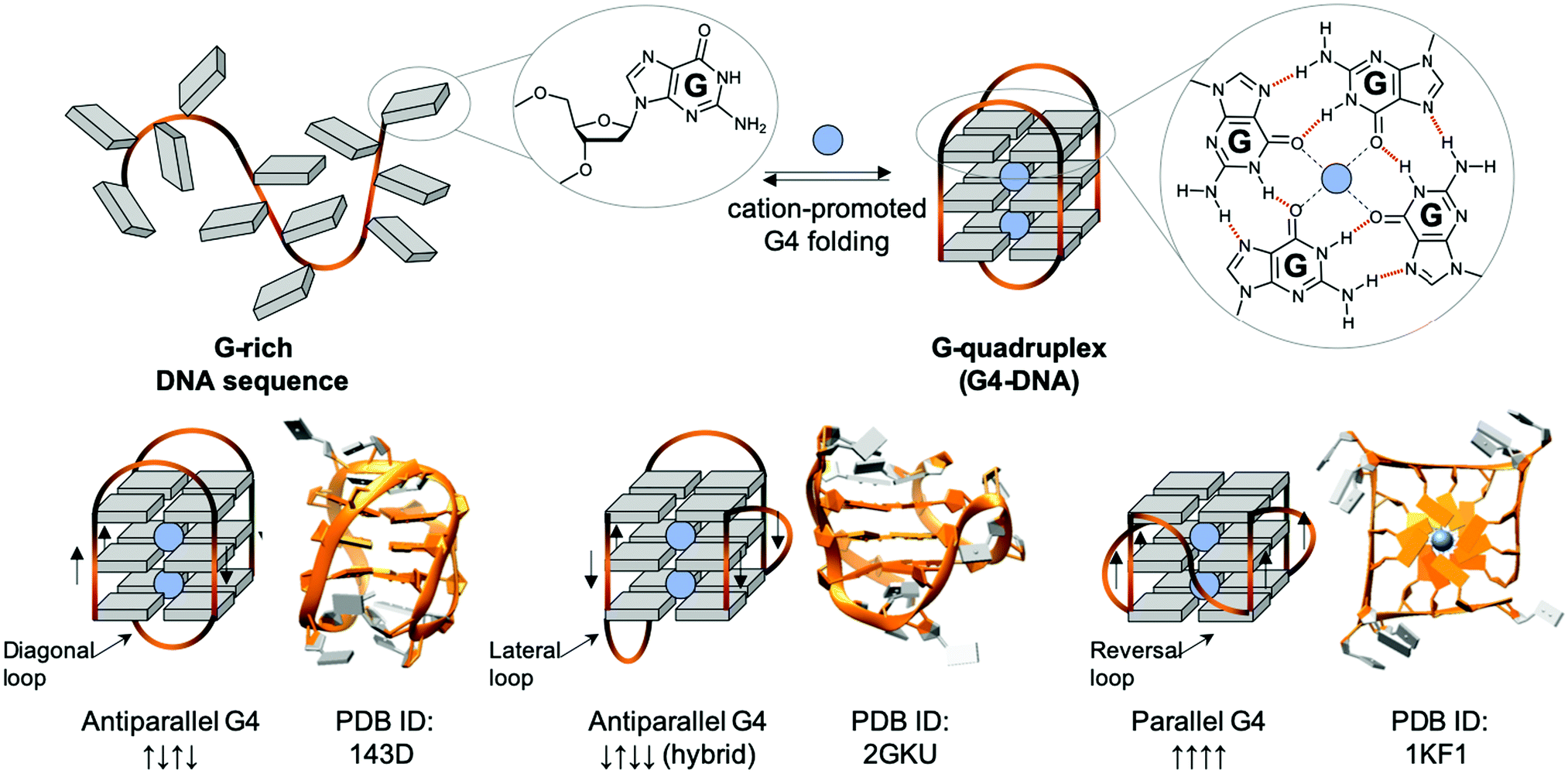 | ||
| Fig. 5 Schematic representation of a G-rich sequence that folds into a G4 structure (upper panel), highlighting the structure of a guanine (G, upper panel, left) and a G-quartet (right). Topological diversity of G4s that can adopt parallel, hybrid and antiparallel conformation (lower panel, arrows indicate the polarity of the DNA strands), as elucidated by either NMR (PDB IDs 143D176 and 2GKU177) or X-ray structure analysis (PDB ID 1KF1178). | ||
G4s in gene promoters: the occurrence of QFS in gene promoters (defined as 1 kb upstream of the transcription start site, TSS) is significantly high in mammals (>2000),187–189 inferring a regulatory role of G4s in gene expression.190 The genesis of G4 structures is linked to a high transcriptional activity, likely due to physical requirements for the DNA duplex to open and for chromatin structure to be relaxed (euchromatin, as opposed to tightly-bound heterochromatin) thus allowing G4s to fold, but this link also indicates a possible causative role of G4s in the recruitment of transcription factors to euchromatin.184 The observation that G4 sequences are significantly enriched in oncogenes and regions predisposed to amplification in cancers explains and warrants the active search for chemicals (small molecule ligands) that can interact specifically with promoter G4s,191 in order to gain control of processes underlying cancer onset and progression. The textbook example is c-MYC, a transcription factor and proto-oncogene whose protein was considered undruggable owing to its lack of catalytic activity, but for which gene expression can be downregulated by ligand-mediated stabilisation of its G4-containing promoter region.43
G4s in replication origins: in homeostatic conditions, DNA replication origins are regulated by two triggers, transcription and G4 formation.192 Genome mapping showed a correlation between replication origins and G4s in metazoans: >70% of initiation sequences are followed by a QFS, also known as Origin G-rich Repeated Element (OGRE).193 Furthermore, a genome-wide CRISPR-mediated gene editing study showed that by inserting or deleting QFSs, replication origin activity could be increased or decreased respectively,194 demonstrating one of the multiple regulatory roles of quadruplexes in healthy dividing cells. However, the detailed mechanism by which G4s act at replication origins is not yet fully understood.
G4s in telomeres: mammalian telomeres are formed by thousands of non-coding repeats (5′TTAGGG3′/5′CCCTAA3′) and a single-stranded G-rich 3′-tail, known as the 3′ overhang.195,196 The telomeric G4 that folds from the G-rich (5′TTAGGG3′) 3′-overhang is undoubtedly the most intensively studied G4 structure, given that it was the first discovered, and that its 3 TTA loops are flexible enough to give rise to different G4 topologies, as exemplified by the 3 structures seen in Fig. 5 (lower panel), formed in either Na+- (PDB ID 143D176) or K+-rich conditions (PDD IDs 2GKU177 and 1KF1178). Telomeres act as a cap at the end of chromosomes, protecting them from enzymatic degradation. Repeated units are removed after each round of DNA replication and cell division (the so-called ‘end replication problem’),197,198 making telomeric regions the ‘mitotic clock’ of the cell, limiting the number of cell cycle divisions before the onset of senescence.196 Telomeres are thus implicated in both cancer and the ageing process.199,200 It is largely accepted that G4s form in telomere ends,201,202 and the earliest evidence of biological G4 formation was obtained at telomeres. Even if telomeric G4s can either protect telomeres against exonucleases203 or jeopardise telomere organisation by preventing telomeric loop (T-loop) formation and telomerase recognition,204–207 they have been mostly studied as targets for fostering chromosomal fragility in cancers.208
These enzymes are deeply involved in genome surveillance and DNA damage repair (DDR). They are known to resolve higher-order DNA structures formed during DNA transactions (e.g. the 5′ flap intermediate resulting from the processing of Okazaki's fragments during replication, or the DNA:RNA hybrids referred to as R-loops formed during transcription) and structures formed during DDR (e.g. the four-way junction known as the Holliday junction that is the central intermediate of HR). Some examples have already been discussed above: BLM is involved in HR-mediated DSB repair (see Section 2b); XPD mediates NER when embedded in the ternary complex TFIIH (see Section 2a1). Of particular interest here is the prevalence of reports that helicases are inhibited by G4 ligands, likely indirectly through DNA blockages (Table 1).
| Name | Family216,217 | Direct-ionality | Species | Activity (for ligand interaction details, see Section 3c) | Associated human disease | Ref. |
|---|---|---|---|---|---|---|
| BLM | RecQ-like SF2 | 3′ → 5′ | H. sapiens | No preferences for a given subclass of G4 (intra- vs. inter-molecular G4, parallel vs. antiparallel G4s); requires 3′-ss overhang for loading; involved in DNA repair via DSB resection and HR; inhibited by G4-ligands (e.g. BRACO19) | Bloom syndrome | 218–220 |
| Sgs1 | RecQ-like SF2 | 3′ → 5′ | S. cerevisiae | Orthologue of H. sapiens BLM helicase; inhibited by G4-ligands PIPER | — | 221 and 222 |
| WRN | RecQ-like SF2 | 3′ → 5′ | H. sapiens | Wide G4 spectrum; requires 3′-ss overhang for loading; manages replication stress, fork arrest and collapse; inhibited by G4-ligand BRACO19 | Werner syndrome | 219, 220 and 223 |
| RecQ | RecQ-like SF2 | 3′ → 5′ | E. coli | Unwinds both intra- and intermolecular G4s; orthologue in H. sapiens is RecQL4; inhibited by G4-ligand NMM | 224 | |
| DHX36 | DEAH, SF2 | 3′ → 5′ | H. sapiens | Also known as RHAU (RNA helicase associated with AU-rich element) or G4R1 (G4-resolvase 1); unfolds both DNA and RNA G4s; preference for parallel G4s; inhibited by G4 ligands PDS and PhenDC | — | 225–227 |
| Pif1 | Pif1-like SF1 | 5′ → 3′ | S. cerevisiae | Found from yeast to human (hPif1); no preference for a given subclass of G4; requires 5′-ss overhang for loading; inhibited by G4-ligand PhenDC3 | — | 228–231 |
| RTEL1 | DEAH, SF2 | 5′ → 3′ | H. sapiens | Involved in genome stability and telomere integrity; unwinds intra- and inter-molecular G4s; inhibited by G4 ligand TMPyP4 | Hoyeraal–Hreidarsson syndrome | 232 and 233 |
| FANCJ | Fe–S, SF2 | 5′ → 3′ | H. sapiens | Orthologue of nematode DOG-1 helicase (C. elegans); promotes DNA repair via HR on interaction with BRCA1; resolves telomeric G4; inhibited by G4-ligand telomestatin (TMS) | Fanconi anemia | 234–236 |
| DDX1 | Fe–S (DEAD-box), SF2 | 5′ → 3′ | H. sapiens | Unlike FANCJ, DDX1 does not resolve intra- and tetra-molecular G4s (preference for two-stranded G4s); insensitive to TMS treatment | Warsaw breakage syndrome | 236 and 237 |
| XPD | Fe–S (Rad3/XPD), SF2 | 5′ → 3′ | S. acidocaldarius | Unlike FANCJ, XPD does not resolve intra- and tetra-molecular G4s; orthologue in H. sapiens is ERCC2; insensitive to TMS treatment | Cockayne syndrome (for ERCC2) | 236, 238 and 239 |
More recently, certain helicases have been shown to unwind G4s in vitro and evidence is now accumulating that they do so in vivo (Table 1).212,214 For instance, BLM localises at telomeres where it allows for proper replication to occur, resolving telomeric G4, an action that is retarded by G4 ligands (PhenDC3).240 Similarly, BLM-deficiency leads to genomic instability caused by elevated levels of recombination by sister chromatid exchange (SCE) events at G4 sites in transcribed regions of the genome.241 WRN is also found at telomeres where it resolves telomeric G4 to repress chromosomal instability, and interact with the shelterin complex to regulate T-loop formation.242 A pioneering study conducted with Dog-1, the Caenorhabditis elegans homologue of FANCJ, showed that deletions in Dog-1 helicase promotes genetic instability by allowing G4s to act as replication barriers.243 In eukaryotic cells, FANCJ-knockdown (FANCJKD) cell growth is strongly inhibited upon treatment with G4 ligand telomestatin (TMS).235 Similarly, FANCJ-null cells show replication fork slowing upon TMS treatment.244 Genome-wide analysis performed with S. cerevisiae by Pif1-ChIP-qPCR showed an enrichment of G4 motifs at Pif1-binding sites, which are responsible for stalling replication forks and are prone to DNA breakage in Pif1-deficient cells.245 This was further substantiated by the demonstration that Pif1-deficient S. cerevisiae displayed enhanced genetic instability triggered by G4 motifs of different (and controllable) stability, and chemical G4 stabilisation via G4 ligand treatment (PhenDC3).246
Beyond their biological roles, G4-helicases have been implemented as biomolecular tools to study and/or modulate G4 landscapes in cells. For instance, Pif1 has been labelled (Pif1-GFP) to track G4 formation in cells through co-localisation with the clickable G4 ligand pyridostatin-α (PDS-α, visualised following in situ click chemistry, see Section 3c).92 Pif1 has also been overexpressed in cells to reverse G4-mediated genetic instability in S. cerevisiae,247 repair dysfunctions in cancer cells248 and cognitive dysregulations in neurons.249 In the latter two examples, Pif1 overexpression was found to remedy phenotypes of G4-mediated functional impairment induced by G4-stabilising PDS treatments. Similarly, BLM was overexpressed to reverse reactive oxygen species (ROS)-induced G4 (and R-loop) formation at transcriptionally active sites, thus restoring the repair process.250
Since the very first prototypes at the end of the 1990s,254–256 hundreds of G4 ligands13 have been developed to act as both reversible modulators of G4 cellular functions257,258 and as probes to visualise G4s intracellularly (thus confirming their bona fide existence).259 Selectivity for G4s over dsDNA was imperative in the development of G4 ligands. Guidelines for selective G4 ligand design include structural prerequisites of a wide π-surface to enable optimal π–π stacking interaction with the accessible G-quartet of a G4, cationic appendages to interact with the negatively charged DNA backbones, and water-solubilising sidechains.260,261 Structural studies in vitro reveal that most G4 ligands stack above the top G-quartet of the G4.262–264 Chemical investigation and design optimisation are always accompanied by the development of ad hoc biophysical techniques to assess in an ever more precise manner the intrinsic quality (affinity, selectivity) of hundreds of candidates that have yet been evaluated. Such techniques include FRET-melting, fluorescence indicator displacement (FID) assays, ESI-MS and surface plasmon resonance (SPR) investigations.265,266 Rational chemical design is now also complemented by high-throughput screening methods (small molecule microarrays,267 and DNA microarrays268) that assess the interaction of hundreds of ligands with hundreds of G4 targets. Such methods offer great potential for the discovery of new G4-targeting chemical scaffolds, as well as a new methodology to compare G4 specificity, where the intra-G4-selectivity is currently a challenging barrier to application.
The ability of G4 ligands to alter cancer cell proliferation is now firmly established.14,257 However, the precise mechanisms by which they exert their anticancer activity must still be clarified through impartial experimentation and in multicellular conditions. In light of what has been described above, it is tempting, but not necessarily justified, to link the observed global antiproliferative activity of a given ligand to a change on the gene that contains the G4 sequence being targeted, and for which the ligand shows in vitro binding to the truncated oligonucleotide (i.e. oncogene expression modulation for promoter G4s; telomere stability for telomeric G4s).190,192,269
And yet, G4 formation is intimately linked to all DNA transactions, favoured by the transient formation of ssDNA during DNA-replication and DNA-to-RNA transcription, when the B-helix is split apart and the single strands are subjected to negative supercoiling, in which helical tension is theorised to cause the DNA to curl like a twisted string. Whilst transactions induce their formation, G4s can physically impede these two processes, stopping or stalling the advancing polymerase, triggering DNA damage and activating DDR mechanisms. This way to consider G4 as a bulky steric hinderance creating DNA damage was – and still is – not well accepted in the field since it removes G4 from its position as a unique genetic lever able to control complicated cellular processes with exquisite spatiotemporal specificity (Fig. 6). However, this approach offers a model that accounts for most if not all results collected in cellular proliferation studies reported so far. Amongst the myriad of candidates synthesised to date, only a handful of ligands have been thoroughly characterised for their ability to trigger DNA damage, and for which the therapeutic potential through DDR modulation has been exploited.
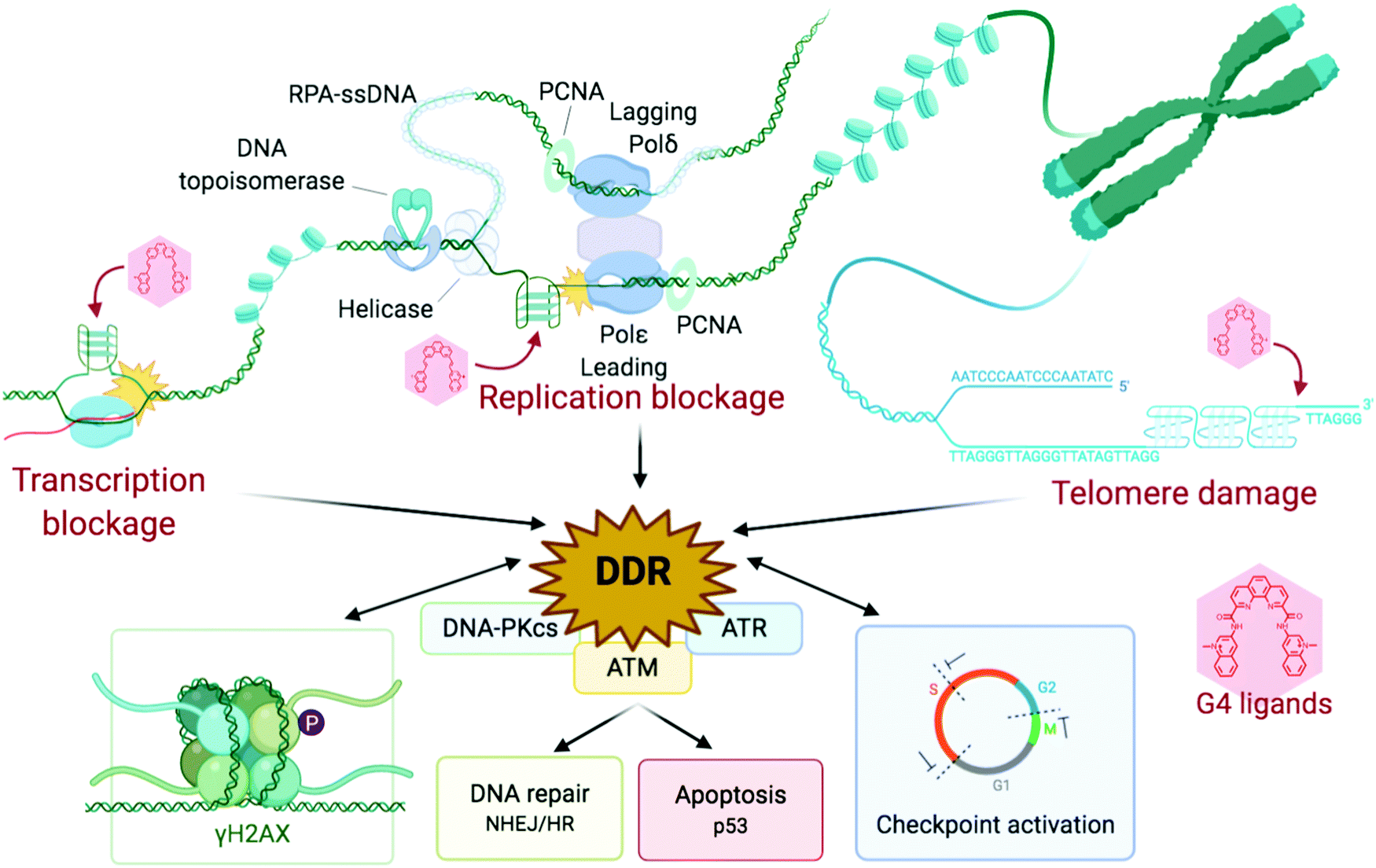 | ||
| Fig. 6 Schematic representation of the stabilisation of G4s, inducing replication and transcription blockages and telomere damage, producing a DDR response, which feeds back into damage signalling (γH2AX) and checkpoint inhibition, and eventually leads to recombination repair and/or cellular shutdown through apoptosis. Created with BioRender. | ||
Telomestatin (TMS): one of the first G4 ligands to be studied in cells was the Streptomyces anulatus-derived natural product TMS, isolated from wide range screening of telomerase inhibitors (chemical structures in Fig. 7a).270 A structural analysis (NMR) of a TMS/G4 complex confirmed that TMS interacts with G4 mostly via π-stacking atop the accessible G-quartet of the telomeric G4.262 When treated with TMS, leukaemia cells proliferate for the first 20 days and apoptosis occurs after this lag (over ten more days), accounting for the delay of telomerase-overexpressing immortal cancer cells to suffer TMS-induced telomerase inhibition and remortalisation (progressive shortening of the telomere leading to chromatin damage and apoptosis, involving activation of p53 and CHK2).271 TMS exerts its activity beyond telomerase inhibition, either disrupting telomere organisation (uncapping telomere-associated proteins),272 or inducing DNA damage (quantified by immunodetection of γH2AX) in both telomeric (telomere dysfunction-induced foci, or TIFs) and non-telomeric regions, which triggers cell cycle arrest in G1 phase and apoptosis.273,274 TMS was also found able to inhibit the expression of genes containing QFS in their promoters (e.g. c-Myb in glioma cells),275 presumably by blocking polymerase processivity. The only sour note of TMS is its complicated accessibility; to tackle this issue, TMS derivative 6OTD was designed (Fig. 7a), as it is more readily synthesised than TMS.270,276 6OTD equally shows interesting G4-interactions in vitro277 and inhibits the proliferation of glioma stem cells via both telomeric and non-telomeric DNA damage, G1-cell cycle arrest and apoptosis.
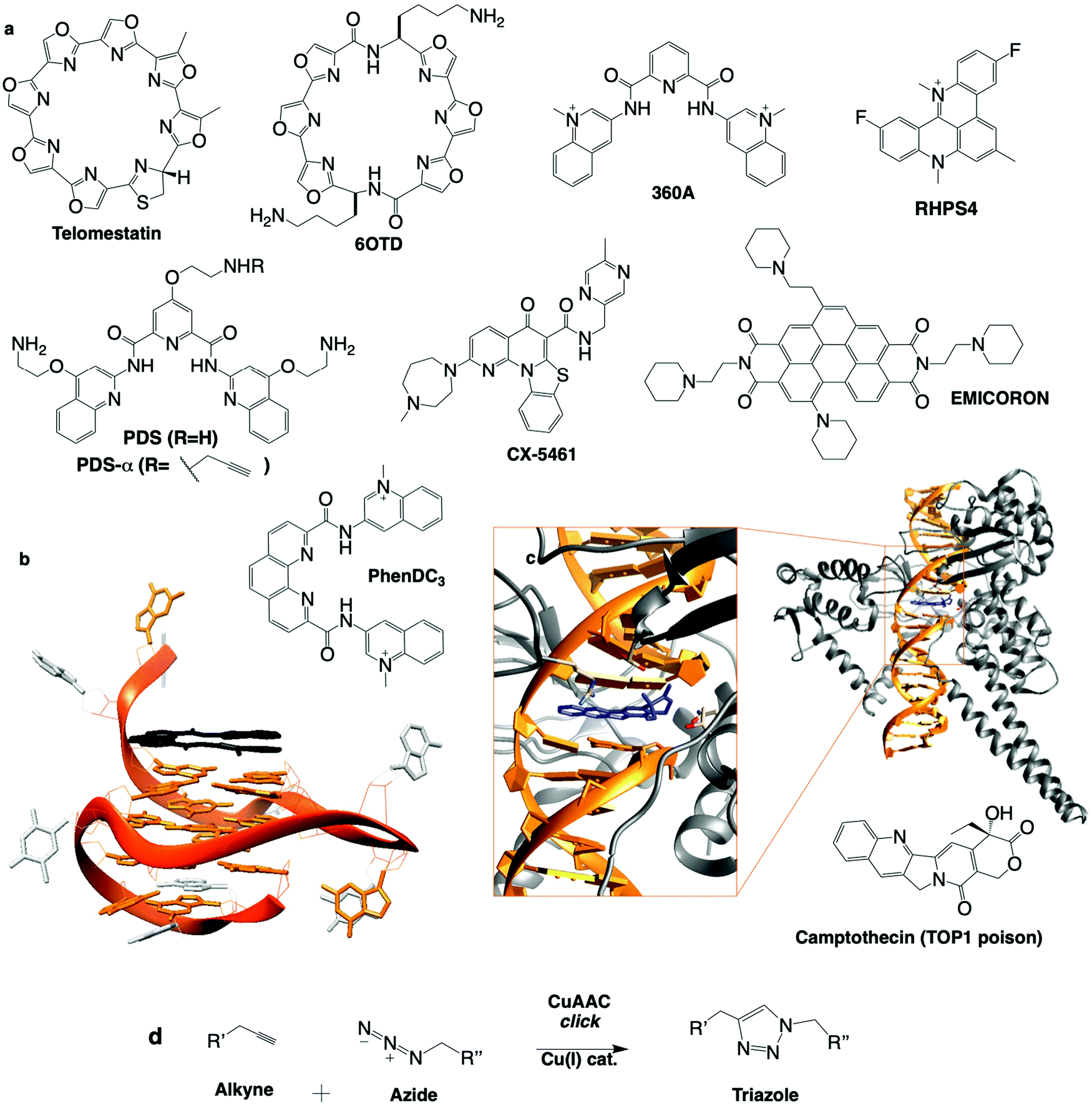 | ||
| Fig. 7 (a) Chemical structures of G4 ligands. (b) NMR structure of G4 with PhenDC ligand (PDB ID: 2MGN).263 (c) Crystal structure of TOP1cc with camptothecin (PDB ID: 1T8I).311 (d) Bioorthogonal copper catalysed click ligation performed in cells between PDSα and an azide-labelled fluorophore. | ||
RHPS4: the pentacyclic acridinium salt RHPS4, whose NMR structure in complex with a G4 has been established,278 has been instrumental in the discovery of G4 ligand-mediated DNA damage. Six human melanoma lines treated with RHPS4 showed an increase in telomere dysfunction and cell cycle inhibition,279 and telomeric DNA damage (TIFs),280 demonstrated by foci of γH2AX, 53BP1 and RAD17 colocalised with shelterin component telomeric repeat factor 1 (TRF1). Analysis of ATRKD and ATMKD cells and with the use of the ATR inhibitor caffeine revealed that RHPS4-induced DDR signalling is dependent on ATR, but not ATM, despite both being activated in WT cells, positioning ATR upstream of ATM in the telomeric DNA damage signalling cascade (Table 2).280 CHK2 was also activated upon RHSP4 treatment and p53/p21 found to be the principal transducers inducing cell death in the same cell line.281 In the same study, RHSP4 was shown to induce damage preferentially in replicating cells, where damage signalling was largely ATR-mediated, indicating its mechanism is carried out by replication fork stalling through stabilisation of G4 structures. This was evidenced by a significant colocalisation of DNA damage marker γH2AX with replication marker PCNA, whereas ionising radiation (IR) induced γH2AX foci independent of PCNA.281 Additionally, helicase activity (WRN and BLM) increased upon RHSP4 treatment, and BLMKD cells showed an increased DDR response to RHSP4-induced damage, further confirming that RHSP4 elicits its damage-inducing activity through the stabilisation of G4 structures.
| Ligand | Sensitising genetic change (knockout/knockdown) | Mechanism implicated | Marker | Cell type/model | Ref. |
|---|---|---|---|---|---|
| TMS | Helicase FANCJKD | Apoptosis | γH2AX | HeLa adenocarcinoma cells, telomerase-negative U2OS osteosarcoma cells | 235 and 312 |
| PDS, CX-5461 | Helicase FANCJKD | Replication stress | HeLa cells | 313 | |
| RHPS4 | ATRKD | Telomere damage | γH2AX, Rad17 and 53BP1 colocalisation with TRF1 | Breast and colon carcinoma-derived mice xenograft | 280 |
| PhenDC | Pif1KO; KO of Rad51, Rad52 and Rad54 desensitises cells | Chromosomal rearrangements | Yeast Saccharomyces cerevisiae | 229, 246 and 288 | |
| TMS | ATRXKO | DNA damage, apoptosis | γH2AX | Murine neuroprogenitor cells | 314 |
| 360A | ATMKD, ATRKD, RAD51KD, DNA-PKcsKD | Telomere damage and dysfunction, | γH2AX/TRF1 colocalisation, CHK2, p53, RAD51, DNA-PK | HeLa, HCT116 colorectal carcinoma, SV40-immortalised As3wt2 human fibroblasts | 284–286 |
| PhenDC | Helicase BLMKD | Induction of subtelomeric origins, replication fork slowing, increased G4 in telomeres | BG4 antibody staining colocalised with TRF1 | SV40-immortalised mouse ear fibroblasts | 240 |
| PDS, PhenDC | RAD51KD, BRCA1−/−, BRCA2−/− (PDS only) | PARP1 cleavage, apoptosis, cell death | γH2AX | MEF fibroblasts | 289 |
| CX-5461, CX-3543 (PDS) | BRCA2KO, BRCA1KD, (BRCA2KD in U2OS), DNA-PKKO, LIG4KO | DNA damage, apoptosis | γH2AX, 53BP1, DNA damage (Comet assay), CHK1/2, RAD51 | HCT116 colon cancer cells, BRCA2+,− ovarian, BRCA2+,− breast, U2OS, HCT116-derived and patient-derived xenografts | 304 |
| CX-5461, PDS | TDP2KO | Inhibition of TOP2cc repair | RPE-1 fibroblasts | 79 | |
| PDS | DNA-PKcsKO | Inhibition of NHEJ-dependent DNA repair | DNA-PK−/− MO59J vs. DNA-PK+/+ MO59K glioblastoma cells | 92 and 315 | |
| PDS | XPFKO/KD and FANCMKO/KD (HR/DSB repair mediators) | Stalled replication forks (DNA fibre analysis) | γH2AX, CHK1 and RPA2 | U2OS | 316 |
| PDS | PolqKO,KD (Polθ protein is involved in alternative end joining DSB repair) | Chromosomal rearrangements, cell death | MEF | 317 |
360A: the pyridodicarboxamide 360A displays a high affinity for G4 and exhibits strong telomerase inhibition in vitro.282 The tritiated 360A (3H-360A) was found to localise at telomeres in T-cell lymphoblatic peripheral blood lymphocytes and human glioma cells by autoradiography.283 360A is a telomere-specific DDR-inducing agent for which ATM was first described as an important DDR mediator, since 360A treatment triggers an increase of chromatin rearrangements and telomere damage (TIFs, telomeric γH2AX and 53BP1 foci) in ATM deficient patient-derived cells and in ATMKD HeLa cells compared to ATM-proficient cells.284 Later, it was shown that 360A treatment induced higher levels of telomere aberrations in ATRKD cells compared to wild type, implicating ATR, as well as ATM, in the response to 360A.285 To further investigate the nature of the repair mechanism triggered on 360A-mediated damage induction, both RAD51- and DNA-PK-depleted cells were treated with 360A,286 RAD51 being associated with HR, while DNA-PK with NHEJ (see Section 2b). Both Rad51KD and DNA-PKKD cells incubated with 360A showed a progressive decline and eventual stop in cell growth. DNA-PKKD was found to inhibit the 360A-induced telomere fusions observed in WT cells, while Rad51KD increased telomere losses and formation of telomere doublets. These results highlight the central role of both HR and NHEJ in 360A-mediated damage repair.
PhenDC: the design of PhenDC3 and PhenDC6 resulted from the structural optimisation of 360A, changing V-shape for U-shape ligands to increase the overlap with the external G-quartet of a G4,287 as demonstrated by NMR (Fig. 7b).263 PhenDC displays tremendous G4-interaction and telomerase inhibition in vitro, sequestering telomere DNA into its G4 fold. PhenDC has been found to protect telomeres against nucleolytic resection by Exo1203 and to trigger genetic instability in budding yeast as a result of Pif1 helicase inhibition.229 A later study by the same group indicated that genetic instability resulted from disrupted HR repair mechanisms (misaligned synthesis-dependent strand annealing reactions), and G4-mediated large-scale genomic rearrangements (in a recombination-dependent manner involving RAD51, RAD52 and RAD54) occurring when G4s fold on the leading strand.288 Similar observations were reported in human cells where PhenDC counteracts helicase processivity (BLM and WRN) and decreases replication fork speed, particularly at telomeres,240 as well as triggering DSBs upon loss of PARP3, which acts as a promoter of chromosomal rearrangements primarily occurring via NHEJ.114 PhenDC treatment of BRCA1-, BRCA2- and RAD51-depleted cells leads to apoptosis, further confirming its capacity to jeopardise HR-mediated DSB repair.289Via an unbiased genome-wide short hairpin RNA (shRNA) genetic silencing screen, a series of PhenDC sensitiser genes was identified (many were in common with PDS treatment, see Table 2), chief among them BRCA1 and TOP1.290
Pyridostatin (PDS): the pyridodicarboxamide PDS displays exceptional in vitro binding properties.92,291 Initially designed as a telomere-targeting agent, it was rapidly shown that PDS targets both genomic G4s (2–5 μM) and telomeric G4s (>5 μM).92 PDS treatment triggers DNA damage, evidenced by phosphorylation of H2AX, and ATM/ATR pathway activation.92,289 PDS stabilises G4 during both transcription and replication, causing DNA and RNA polymerase stalling.92,292 DNA damage induced by PDS is repaired in both a NHEJ- and HR-dependent manner.92,289,293 PDS treatment of BRCA2KD or RAD51KD cells showed robust activation of CHK1 and RPA thus implicating ATM/ATR signalling. More recently, independent non-biased genetic approaches showed that the cytotoxic effects of PDS (and CX-5461, see Table 2) are dependent on TOP2 activity suggesting that PDS could trap TOP2 at G4.79,91 Beyond its therapeutic properties, PDS is also an exquisite molecular tool to study G4 both in vitro and in vivo. For instance, PDS is widely used to favour G4 formation in cell lysate for G4-seq and rG4-seq sequencing-based techniques.258 Several derivatives of PDS have been designed and used to assess the relevance of G4 in cells. For instance, a biotinylated PDS was used to pull down G4s from cell extracts,294 and an alkynylated PDS (PDS-α) was used to localise G4s directly in the nucleus by fluorescence microscopy after copper-catalysed alkyne azide cycloaddition (CuAAC) click ligation with a fluorophore in fixed cells92 (Fig. 7d). This bioorthogonal technique to visualise a small molecule's cellular localisation in situ is particularly precise due to its non-perturbing nature;295 the small molecule of interest with only a miniscule chemical modification binds to its biological target, and can be observed following specific conjugation with the clickable fluorophore, thus introducing the least experimental bias possible. This was a cornerstone study as PDS-α-binding sites were found to significantly co-localise with γH2AX and only minorly with telomeric TRF1, indicating that PDS induces DNA damage primarily at genomic G4 sites. Beyond building the link between G4s and DNA damage, this approach also provided the very first description of the G4 landscape within human cells (via an accurate mapping of the distribution of G4s in the endogenous chromatin context) and highlighted the druggability of the SRC proto-oncogene (involved in multiple pathways that regulate tumour progression),296 thus uncovering a novel G4-mediated anticancer strategy. Recently, a PDS analogue conjugated to a fluorophore (SiR-PyPDS) was used in single-molecule live-cell fluorescence imaging to track dynamic G4 formation,185 demonstrating the density and lifetime of G4s, and showing a significant increase in G4s in cells undergoing transcription and replication compared to steady state (G0/G1) cells. An alkynylated PDS derivative was chemically ligated to a ICL-inducing nitrogen mustard chlorambucil (Chl) (by CuAAC in vitro) to form a G4 ligand-cross-linking conjugate293 (Fig. 7d). This PDS–Chl drug conjugate showed a greater specificity for NER-deficient cancer cells compared to each of the two drugs alone. Such G4-targeted drug conjugates could be further exploited in NER-related diseases such as skin, testicular and drug resistant cancers. Finally, PDS has been used in non-cancer studies, to demonstrate a G4-mediated down-regulation of BRCA1297 and Autophagy related Gene (ATG7) in neurons,249 thus linking G4s to ageing and neurodegenerative diseases.188
CX-5461: fluoroquinolones are a group of molecules described to have topoisomerase II (TOP2) and G4 binding affinities.191,298 Structural optimisation of fluoroquinolones to improve G4 versus TOP2 affinity first gave rise to CX-3543 (also known as quarfloxin), the first G4-interacting ligand to reach clinical trials. The described mechanism of action involves the disruption of nucleolin binding to G4-containing ribosomal DNA resulting in the inhibition of RNA polymerase 1 (Pol1) (overexpressed in tumours due to their increased need for protein production) leading to cancer cell death.299 CX-5461 was designed subsequently and described again as a Pol1 inhibitor although with no mention of G4 interaction in publications from Cylene Pharma.300,301 In an independent study, CX-5461 was later shown to induce a DDR response (ATR/ATM activation) and G2 phase cell cycle arrest in leukaemia cells.302 This unexpected behaviour was later correlated with its ability to stabilise G4s and stall replication forks.303,304 In this study, the ability of CX-5461 to interact with G4s was established both in vitro (via FRET-melting assay) and in vivo (with a significant increase in the G4 nuclear foci labelled with the G4-specific antibody BG4).305 BRCA2-depleted cells were found far more sensitive to CX-5461 treatment compared to WT cells, supporting an impact of G4 ligands on replication forks and implicating HR in the repair of the resulting structures. Chromatin immunoprecipitation (ChIP) of RAD51 and ChIP-seq studies performed in U2OS cells supported that some of the DNA damage caused by CX-5461 is repaired by RAD51.304 NHEJ is also involved in the response to CX-5461 as a hypersensitivity to CX-5461 was also observed in DNA-PKKD cells. However, recent unbiased genetic approaches supported that both CX-5461 and PDS trigger DNA damage and mediate their cytotoxic effect through TOP2 interaction more specifically with TOP2A.79,91,306 These studies provide mechanistic insights into how CX-5461, PDS and potentially other G4 ligands produce DNA damage through structure specific interference with DNA topoisomerases.
To conclude, these six ligands are illustrative examples of G4 ligands that trigger DNA damage with varying ability, leading to multiple and varying cellular responses, all of them with clinical potential. A more comprehensive view of the genetic modifications described to sensitise cells to G4 ligands is given below, and illustrates the extreme correlation with DDR pathways (Table 2). New ligands are regularly reported (e.g. SYUIQ, 20A and FG) with yet again slightly different DNA damaging specificities.307–309 All of these chemicals target G4 in vitro and in cells, and only with the help of non-biased analyses,79,91,290 will we decipher how stabilisation of these structures translates into phenotypes that could be exploited to cure diseases. Surprisingly or not, topoisomerase inhibition (TOP1/2) is often evoked, leading one to consider that G4 ligands could trap DNA-bound TOP1 or TOP2 complexes in a structure-specific manner. TOP1/2 are responsible for untwisting DNA under torsional strain via formation of a topoisomerase–DNA cleavage complex.58 Topoisomerase poisons are commonly used anti-cancer agents (see Section 1b). They behave as interfacial inhibitors inserting into the topoisomerase–DNA complex and thereby blocking the religation step of the topoisomerase catalytic cycle and stabilising the cleavage complex (cc, TOP1cc or TOP2cc) (Fig. 7c). This is exemplified by prototype anticancer agents camptothecin (TOP1 poison, active site crystal structure in Fig. 7c),310 doxorubicin and etoposide (TOP2 poisons).60
The advantage that G4 ligands would then have over classical topoisomerase inhibitors is a double specificity, for a structure of nucleic acids and for regulatory sequences in which G4 forming sequences are more prevalent. It is to be noted that topoisomerases accumulate at sites of DNA aberrations,62 and maybe even at G4s. Only further studies will help gain insights into the actual targets of G4 ligands in cells, with the hope of gaining “a deep understanding of cell circuitry and diseases biology” that the G4s are involved in.
Mechanistic studies frequently describe a (hyper)sensitisation (increased growth inhibition) of cancer models to G4 ligands, when comparing the DDR-deficient cells/tissue with the wild type (Table 2) or when cells/tissue are cotreated with antiproliferative agents or DDR inhibitors (Table 3). Exploiting DDR-deficient cancer cells has been shown for BRCA1/2-deficient cells, which are sensitised to G4 ligands (PDS,315 PDS and PhenDC289) compared to BRCA1/2-proficient cells and one molecule, CX-5461, is in clinical trials for BRCA1/2-deficient tumours.304 PDS and CX-5461 exacerbate the damage occurring in DDR-deficient cells, similar to PARP inhibitors. In the same system, the damage-inducing effect was amplified by cotreatment with a DNA-PK inhibitor NU7441.315
| Ligand | Synergistic DDR drug/treatment | DDR mechanism implicated | Marker | Cell type/model | Ref. |
|---|---|---|---|---|---|
| TMS | Imatinib, daunotubicin, mitoxantrone, vincristine | Telomere damage | CHK2, ATM, p21, p27 | K562, OM9;22 leukaemia cells | 271 |
| TMS | NSC19630 (WRN helicase inhibitor) | Stalled replication, DNA damage, cell death | γH2AX, PCNA | U2OS | 318 |
| RHSP4 | Taxol | Telomere damage, chromosome fusions | γH2AX/TRF1 | UXF1138L cells and xenograft | 319 |
| RHPS4 | Camptothecin (TOP1 inhibitor) then RHPS4 (CI < 0.2), bleomycin (CI ≈ 0.7) | Apoptosis, tumour growth inhibition | γH2AX | M14 melanoma cells, HT29 colon-derived xenograft | 320 |
| RHPS4 | GPI 15427 (PARP inhibitor) followed by RHPS4, camptothecin | Telomere damage chromosome aberrations (or PARP1KD sensitisation) | PARP/telomere colocalisation | BJ fibroblasts, HT29-derived xenografts | 321 |
| RHPS4 | SN-38 or ST1484 (TOP1 inhibitors) followed by RHPS4 (CI ≈ 0.1) | Telomere damage, cell death | γH2AX, TOP1 accumulation at telomeres | BJ fibroblasts, HT29 | 322 |
| No synergy with adriamycin and doxorubicin (TOP2a inhibitors) | |||||
| RHSP4 derivates | SN-38 (CI < 0.5) | Telomere damage, stalled replication | γH2AX/TRF1, PCNA | HT29 | 323 |
| RHPS4 | Ionising radiation (IR) | Telomere damage, G2-phase block, chromosomal rearrangements, tumour growth inhibition | γH2AX and 53BP1/TRF1 colocalisation | U251MG glioblastoma cells and xenograft | 324 and 325 |
| PDS | NU7441 (DNA-PK inhibitor) | Induction of DNA:RNA hybrid structures (R-loops), DNA damage, DSBs and DDR signalling | γH2AX, 53BP1, ATM, antibody colocalisation: BG4 (G4) and S9.6 (R-loop) | DNA-PK−/− MO59J vs. DNA-PK+/+ MO59K glioblastoma cells | 309 |
| PDS, FG | NU7441 (DNA-PK inhibitor) | DSBs, ATM-dependent DDR | γH2AX, ATM | SV40-transformed MRC-5 fibroblasts | 92 |
| PDS, PDSI | NU7441 (DNA-PK inhibitor) | DSBs, DDR, apoptosis | 53BP1, RAD51 (or BRCA2−/−) | HCT116 colon cancer cells | 315 |
| PDS | MK1775 (WEE1 cell cycle kinase inhibitor) and pimozide (FA repair USP1 inhibitor) | G4 stabilisation, cell death | Sensitised to KD of BRCA1&2, POLQ, USP1, TOP1, WEE1 etc. | A375 melanoma cells, HT1080 fibrosarcoma cells | 290 |
| PhenDC | G4 stabilisation, cell death | Sensitised to KD of BRCA1 and TOP1 | |||
| PDS, PhenDC | ME0328 (PARP3 inhibitor), KU0058948 (PARP1 inhibitor) | Chromosome rearrangements, DSBs, G2-phase block | γH2AX, 53BP1, G4-binding 1H6 antibody staining, PARP3KO sensitised | A549 lung adenocarcinoma cells | 114 |
| CX-5461, CX-3543. PDS | IR, hydroxyurea | DNA damage | γH2AX, 53BP1, CHK1/2, RAD51, ATRXKD sensitised | Murine normal human astrocytes, glioma stem cells | 326 |
| CX-5461 | BMN-673 (PARP inhibitor) | Pol1 transcription inhibition, replication stress, DNA damage, G2-phase block, no G4-stabilisation effect | ATM/ATR, RPA, γH2AX, HR-deficient, BRCA1−/− and MYC−/− cells sensitised | Ovarian cancer cells and xenografts | 303 |
| EMICORON | SN-38 then EMICORON; EMICORON then oxaliplatin or 5-FU (all CI < 0.5) | Telomeric DNA damage | HT29 colon cancer cells and xenografts | 327 |
One of the earliest studies on TMS investigated the combinatorial effects with multiple antiproliferative agents.271 Tauchi et al. described an additive effect of TMS with imatinib (inhibitor of anti-apoptotic tyrosine kinases), daunorubicin (TOP2 inhibitor), mitoxantrone (intercalator and TOP1 inhibitor) and vincristine (microtubule inhibitor), analysed by cell growth inhibition in single cell culture (Table 3). Several studies have exploited the telomere selectivity of RHPS4 to induce anti-tumoral activity. Such interactions are often analysed by colocalisation of γH2AX damage signalling with telomere marker TRF1, particularly in the case of RHPS4 and 360A, which exhibit various synergistic and sensitising interactions. WRN inhibitor NSC-19630 was found to induce replication-dependent DNA damage (γH2AX/PCNA colocalisation), cell cycle inhibition and apoptosis as a single agent.318 Since G4s are a known substrate of WRN helicase, NSC-19630 was combined with a G4-ligand to enhance these DDR-inducing effects. A non-toxic concentration of NSC-19630 (1 μM, where IC50 = 3 μM) in combination with a non-toxic concentration of TMS (0.6 μM) was shown to induce 70% cell death in 3 days. Still, more data points are required to draw a strict conclusion on synergism.
Synergistic interactions can be confused with additive interactions. To be sure that a combinatorial effect is synergistic, a matrix of concentrations of the two chemical components should be analysed. One popular method pioneered by Chou and Talalay takes into account the shape of each IC50 curve (S-shaped or linear), in order to calculate a combination index (CI), where CI < 1 indicates synergism, CI ≈ 1 indicates additivity and CI > 1 indicates antagonism.328,329 This method was used by Biroccio et al. to show strong synergistic effects between TOP1 inhibitors SN-38 or ST1484 and RHPS4 with extremely low combination indexes (CI < 0.1).322 Another mathematical method to distinguish additive from synergistic effects is the Bliss independence model, in which the calculated additive effects are subtracted from the observed effects to produce a surface plot for a matrix of concentrations.330 In a field in which combinatory drug regimens are standard in the clinic, and clinical trials rarely test every combinatorial option (for a 2 drug combination of A and B, four regimens are possible: ±A/±B), it is crucial to understand the interactions of each drug in fine detail in pre-clinical studies.
Many studies show that G4-induced damage occurs in both the HR and NHEJ pathways,286 and replication- and transcription-dependent mechanisms.92 Due to the vast range of interactions observed for G4 ligands, it is crucial to undertake unbiased experiments, despite the frequent and disappointing discovery of small molecules with poor specificity. CX-5461 was first described as a RNA Pol1 transcription inhibitor via a phenotypic screen, whilst its principal mechanism was more recently shown to be through G4-mediated interference with TOP2.304 Bruno et al. recently showed that CX-5461 incurs resistance to doxorubicin in a colorectal cancer model,306 an important finding because the TOP2 poison doxorubicin is a common treatment for colorectal cancer. CX-5461 is currently in clinical trials for relapse therapy based on the idea that it targets RNA Pol1-dependent transcription.303 However, the recent discovery that it acts through G4-dependent interference with TOP2 should lead to the patient/cancer selection process for this drug being revisited.79,91,306
Advances in genome-wide sequencing techniques such as ChIP-seq and Chem-seq have facilitated the characterisation of genetic and epigenetic modulators.331 A combination of specific DNA targeting agents with chromatin-interacting protein modulation (a field also currently undergoing important drug development innovations)332–334 could prove to be another promising strategy in synthetic lethality chemotherapy.
In light of the wealth of preclinical data acquired with G4 ligands in the past recent years (Table 3), it is now tempting to develop drugs targeting other DNA folds that offer more defined anchoring sites than the external G-quartet. This opens new avenues to develop drugs with higher selectivity and new ways to interfere with DNA transactions in cancer cells. Their relevance to DDR and their therapeutic potential thus merit further investigation, which are highlighted in the following sections.
4. Non-B DNA structures
 | ||
| Fig. 8 Alternative DNA structures form in the vicinity of DNA transactions. The junction point of a cruciform structure presents a four-way junction (FWJ, e.g. Holliday junction) and that of a slipped loop presents a three-way junction (TWJ). Stabilisation of these structures with chemicals (ligands) can impede DNA transactions and induce DDR. Adapted from ref. 35, created with BioRender. | ||
R-Loop structures were first described in 1976347 and currently are receiving renewed attention. R-Loops are DNA:RNA hybrids formed during transcription when the nascent RNA strand folds back and hybridises to the dsDNA sequence it was transcribed from, displacing the DNA strand of similar sequence to form a three-stranded RNA displacement loop, or R-loop.342,348 Initially thought to be formed only accidently, genome-wide analyses such as DRIP-seq349,350 (DNA:RNA immunoprecipitation and sequencing) and DRIVE-seq350 (DNA:RNA in vitro enrichment and sequencing) highlighted the prevalence of R-loops in the human genome, whose distribution implies roles in DNA replication initiation, transcription termination and chromatin patterning.342,348 In a recent study, R-loops generated by RNA polymerase II localised to non-coding regions in ribosomal DNA and had positive regulatory effects on ribosomal RNA expression by RNA Pol1 in the nucleoli.351 Aberrant R-loop formation threatens genomic stability, notably inducing replication and translational stress as they form roadblocks to polymerase processivity that promote DNA breaks and activate both ATM and ATR pathways. However, the relationship between DNA damage and R-loop formation is intricate as R-loops trigger DNA damage and conversely DNA damage favours R-loop formation.352,353 From a chemical biology point of view, R-loops thus represent another class of nucleic acid targets to damage DNA, an approach that has thus far only been attempted indirectly as no R-loop ligands are yet reported. For instance, R-loop formation was promoted by impeding canonical RNA maturation and spliceosome assembly353,354 using the TOP1 inhibitor diospyrin D1,355 or by stabilising G4 with PDS356 and monohydrazone derivatives,357 since stabilised G4s promote the formation of R-loops at transcribed genes and the reciprocal G4/R-loop stabilisation can arrest polymerases and trigger DSBs accumulation.
Inverted repeat sequences are also highly prevalent in the human genome, and are also involved in genetic disorders, they are thus interesting targets for small molecules. Inverted repeats fold into hairpins stabilised by Watson–Crick-type hybridisation of an upstream region with the complementary downstream region. Two types of hairpin structures can be formed: if the inverted repeat-containing sequence forms two hairpins directly opposite to each other, the higher-order structure is referred to as cruciform DNA, or a four-way DNA junction (FWJ); alternatively, if the two hairpins are misaligned (shifted relative to each other), the structure is referred to as slippage loops, S-DNA, or three-way DNA junction (TWJ) (Fig. 8).11,358 The thermodynamic stability of these DNA junctions is high enough to act as physical obstacles that stall the replication fork/transcription bubble, creating replication- and transcription-coupled DNA damage that trigger DDR. Stabilisation of such blockages by external chemicals equally represents an alternative way to trigger DNA damage, with the strategic advantage of presenting structurally defined binding sites (chiefly the central cavity of the DNA junction, often referred to as the junction point) for small molecule targeting with high specificity (see Section 4b).
The quest for DNA junction-forming sequences in our genome has been far less intensive than that of QFS. However, several bioinformatic tools are being developed, such as the Palindrome analyser359 and the IRFinder,360 and preliminary results indicate that inverted repeat-forming sequences might be even more prevalent than QFS. For instance, comparison of the number of inverted repeat-forming sequences and QFS as identified with IRFinder361 and G4-hunter362 algorithms in the genome of S. cerevisiae indicates that there are >8000 inverted repeat-forming sequences (of 30 nt length) versus >3000 QFSs (of 25 nt length), with a strong enrichment at centromeric regions and replication origins. In the human genome, the advent of next-generation sequencing highlighted the prevalence of repeated sequences, accounting for ≈50% of our genome: the most frequent repeats are short and long interspersed nuclear elements (100–300 bp (base pairs) and >300 bp respectively) which account for 36% of the genome (>1500000 sequences), while short tandem repeats (referred to as microsatellites (2–10 bp), minisatellites (10–60 bp) and satellites (60–100 bp)) are adjacent repeating sequences and account for 3% of the genome (>400000 sequences), a percentage comparable with the coding region of the genome.337 However, the proportion of inverted repeat-forming sequences among repeated sequences is less defined: they can be further classified as short and long inverted repeats (<30 bp and >30 bp respectively). Sergei M. Mirkin pioneered the quantification of short inverted repeats in a pre-sequencing era, developing an algorithm that showed that 50% of 40 kbp segments in the human genome contain at least one inverted repeat-forming sequence (with 2 to 6 nt loops).363 Subsequently, Karen M. Vasquez showed that short inverted repeats are significantly enriched at translocation breakpoints (an average of 28.5 short inverted repeats within a ±100 bp region surrounding the 20000 breakpoints, versus 21.4 in 20000 control sequences).364 Long inverted repeats were quantified in a more straightforward manner, with >2500 sequences in the human genome.365
While less studied than G4s, inverted repeats are also known to be responsible for genetic instability, which may have advantageous therapeutic payoffs (see Section 4b). Inverted repeats have been particularly studied in the context of the genetic disorders that hairpin-forming sequences are involved in, known as repeat expansion diseases.11 Indeed, the genetic expansion (in which the sequence gets repeated during replication) of inverted repeats is frequently encountered in both cancers366 and severe neurological disorders (>50 human diseases).11 Repeats are prone to expansion as a result of replication slippage, faulty replication and improper repair.18,367 Efforts have been invested to provide a unified model for explaining expansion (such as the breakage-fusion-bridge (BFB) model368), but the diversity of cellular mechanisms, events and protagonists that could account for repeat expansion explains why the mechanism behind expansion is still unknown. The most common expansion is the trinucleotide repeat expansion369 and related diseases (causative of 16 different inheritable conditions), such as (CAG)n responsible for the Huntington disease, (CGG)n for the fragile X syndrome, and (GGA)n for Friedreich ataxia. The ability of these repeats to fold into hairpin structures has been firmly demonstrated.
As indicated in Section 3b, the cell has evolved the helicase machinery to resolve branched DNA structures219,370 which might be formed during DNA transactions and repair. Inverted repeats were significantly more present at replication fork stall sites in E. coli, S. cerevisiae and monkey fibroblasts cells, in a hairpin length-dependent manner.371,372 In another study, a non-biased RPA-ChIP-Seq sampling of replication fork stall sites reported that inverted repeats and structure-forming (purine-rich and AT-rich) microsatellite sequences were enriched at fork stall sites.373 This effect was amplified by chemical ATR inhibition, further indicating that the DDR machinery is used to straighten out these inverted repeat structures in homogenous conditions. The instability triggered herein is also counteracted by fork-stabilising proteins topoisomerase 1-associated factor 1 (Tof1) and Mannose Receptor C-Type 1 (Mrc1), that prevent irreversible fork collapse and DNA breakage.371,374 Several DDR mechanisms have been implicated in inverted repeat-induced damage and repair. Short inverted repeats were also shown to trigger genetic instability via DSB formation after being cleaved by the MRX (MRE11-RAD50-Xrs2 in S. cerevisiae, MRN in human) complex associated with Sae2 (SUMO1 activating enzyme subunit 2, CtIP in human)375 and repaired by HR,376 or cleaved by the ERCC1–XPF complex364 and repaired by the microhomology-mediated end joining (MMEJ) mechanism.377 These results explain why targeting inverted repeat-forming sequences with chemicals fosters lethal DNA damage and trigger DDR (Fig. 8).
 | ||
| Fig. 9 (a and b) Crystal structure of FWJ with bisacridine ligand (PDB ID: 2GWA).378 (c and d) Crystal structure of TWJ with supramolecular iron cylinder (PDB ID: 2ET0),379 (e) FWJ ligand WRWYCR peptide in active disulfide form, TWJ ligands azacryptand TrisPOB (f) and azacyclophane 1,5-BisNP-O (g). | ||
The biological relevance of targeting inverted repeats was first demonstrated with the studies of peptidic ligands that bind the junction point of FWJs. In vitro and in E. coli, hexapeptides were capable of selectively binding the FWJ of a Holliday junction (a crucial intermediate during HR repair of both DSBs and replication-dependent ssDNA gaps, as demonstrated by live cell imaging and HJ-ChIP-seq),405,406 thus abrogating the action of Holliday junction-processing enzymes by displacing RecG helicase and inhibiting RuvABC resolvase, in the case of WRWYCR, for which the active form is the disulfide-bridged dimer (Fig. 9e).388,389 Furthermore, a similar more selective hexapeptide WKHYNY, triggered the accumulation of DNA breaks by blocking DNA repair through the inhibition of Cre recombinase,407 and was shown to act synergistically with other DNA damaging agents such as cross-linking agent mitomycin C.408 Later on, these peptides were shown to inhibit proliferation of numerous cancer cell lines (PC3, Du145, LnCAP, DuPro-1, PPC-1, HeLa and A549) via DNA damage induction, demonstrated by the accumulation of γH2AX and 53BP1 foci and activation of CHK1 and CHK2.409 Again, the antiproliferative activities of these peptides were amplified by DSB-inducers (TOP2 inhibitors doxorubicin and etoposide) but not by unrelated therapeutics such as the mitotic inhibitor docetaxel, lending further credence to the DNA repair-based therapeutic activity of FWJ-ligands.
In these approaches, FWJ-targeting agents were used to impair DNA repair and their cellular activity is potentiated by DNA damaging agents. Similarly, in a more recent study, TWJ-targeting agents were shown to induce DNA damage, and their cellular activity potentiated by DNA repair inhibitors. The use of TWJ-targeting was greatly aided on obtaining the crystal structure of a supramolecular iron cylinder nestled into the junction point of a TWJ379 (Fig. 9c and d). This structure offers a brand new antiproliferative approach based on the hypothesis that an “iron cylinder is able to bind to and stall DNA replication forks (a three-way junction structure)”,410 a hypothesis not entirely accurate given that replication forks are not TWJ structures per se. Despite being able to disrupt DNA transactions in vitro,411 these compounds were found to be poorly toxic in both cancer (HBL100, SKOV3, T47D and HL-60) and non-cancer (MRC5) cells, unable to trigger DNA damage, but capable of driving cells towards apoptosis.410 Alternative investigations questioned both their TWJ-specificity412–416 and even the DNA-interacting properties of this class of ligands.417
These results created momentum for further development, notably via the studies of cryptand-type ligands of two types: azacyclophanes, also known as cyclo-bis-intercalators; and azacryptands, also known as cyclo-tris-intercalators (Fig. 9f and g). These ligands, initially designed to target mismatched base pairs418,419 and abasic sites,420,421 have a 3D structure fully suited to fit within the TWJ junction point.400 The TWJ-interacting properties of these ligands were first thoroughly assessed via a series of in vitro assays (TWJ-screen, FRET-melting assays, etc.)401,402 to select candidates displaying high affinity and selectivity for TWJs, in order to provide a reliable bona fide model for interpreting cellular studies. The best candidates displayed antitumoral activity in the low micromolar range against MCF7 and MDA-MB-231 cells402,422 due to their ability to create DNA damage (γH2AX accumulation) mostly at the G1/S transition. The TWJ ligands were then used in drug combinations with DDR inhibitors, targeting both HR and NHEJ: NU7441 (DNA-PK inhibitor which impairs NHEJ, structure in Fig. 4b), KU55933 (ATM inhibitor which impairs DSB signalling and HR) and B02 (RAD51 inhibitor which impairs HR). Combination indexes (CI) as low as 0.39 (B02), 0.72 (KU55933) and 0.80 (NU7441) demonstrated that these azacryptands trigger both transcription- and replication-associated DSBs. The excellent synergy (CI = 0.39) obtained with azacryptand TrisPOB (Fig. 9f) and RAD51 inhibitor B02 supports that TWJ-ligands induce DSBs repaired by HR, potentially by promoting fork stalling in S and G2 phase.6,16
Conclusion
Francis H. C. Crick, one of the founders of modern genetics, the co-discoverer of the double helix of DNA (1953)26 and the father of the central dogma of biology (1957),423,424 wrote in 1974: “we totally missed the possible role of enzymes in repair although (…) I later came to realise that DNA is so precious that probably many distinct repair mechanisms would exist”.425 Because of this oversight, DNA repair has required a long time to be discovered, studied and understood. This lag also stems from the multiplicity of pathways involved in DNA repair thoroughly described above, along with their intricate cross-talk. As a consequence, a therapeutic approach that aims at damaging DNA is far more challenging than anticipated, as repair takes place under many different guises and innumerate feedback mechanisms to optimise its efficiency.While it has been attempted to target the DNA double helix directly, we highlight here that DNA is a highly dynamic molecule that can adopt many higher-order structures, which represent a new class of targets to inflict severe injuries on cancer cells’ genome. The targeting of these structures can be performed with good-to-excellent specificity (as compared to the targeting of B DNA) and it has now been firmly demonstrated that the stabilisation of these structures by small molecules triggers extensive DNA damage that ultimately leads to cell death. Here, we focus on how damage is triggered in cells and describe the many different signaling and repair pathways that are activated in response to non-B DNA structure-mediated DNA damage.
Pioneering studies using G4 ligands have paved the way for understanding how the stabilisation of G4s creates damage and the therapeutic utility this presents in cancer. New pre-clinical and clinical data is emerging on the use DNA damaging agents (G4 ligands) with inhibitors of DNA repair, an approach referred to as combination therapy or chemically induced synthetic lethality. It is of unquestionable importance to perform detailed mechanistic investigations, and the recently developed, non-biased approaches such as chemoproteomics, chemogenomics and functional genomics are invaluable. Only an in-depth understanding of the damage caused by targeting alternative DNA structures can help define them as new biomarkers for cancers therapies.
The recent development of ligands which recognise other alternative DNA folds (such as three- and four-way DNA junctions) are of immense value in determining their prevalence in cells, understanding their functions and assessing their relevance as therapeutic targets. Although in its infancy, this strategy benefits from the momentum provided by G4 studies (in terms of tools and strategies, techniques and assays, etc.) and, importantly, opens brand new therapeutic opportunities and chemical space. The recent demonstration that the stabilisation of DNA junctions triggers DNA damage enables the design of new therapeutic cocktails, combining ever more specific DNA damaging agents and DNA repair inhibitors, to halt cancer cell proliferation in a synergistic manner. Efforts must now be invested to validate this strategy at the pre-clinical/clinical levels, which will represent long but worthwhile efforts because, as cancer is a major pressing societal concern, all opportunities are valuable.
The examples depicted above show that alternative nucleic acid structures have gone a long way since their discovery, from being considered in vitro artefacts to biologically functional and targetable entities. These advances have been made possible thanks to an ever deeper understanding of the cell's inner workings which results from decades of resolutely multidisciplinary research combining molecular and cell biology, chemistry and biophysics. These results thus are a perfect demonstration of the absolute necessity to work at the interface between different, strongly interdependent scientific disciplines to enable science to meet societal expectations for the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Centre National de la Recherche Scientifique (CNRS), the Agence Nationale de la Recherche (ANR-17-CE17-0010-01), the Université de Bourgogne, Conseil Régional de Bourgogne and the European Union (PO FEDER-FSE Bourgogne 2014/2020 programs, LS 206712) and the INSERM Plan Cancer 2014–2019 (19CP117-00) for financial support. The authors also thank all scientists worldwide involved in these fascinating fields of research, to make it lively, thrilling and always moving, and apologize to all colleagues whose important contributions could not be cited owing to space limitations.References
- D. Hanahan and R. A. Weinberg, Cell, 2000, 107, 57–70 CrossRef.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS.
- S. P. Jackson and J. Bartek, Nat. Rev., 2009, 461, 1071–1078 CAS.
- M. J. O’Connor, Mol. Cell, 2015, 60, 547–560 CrossRef.
- A. Minchom, C. Aversa and J. Lopez, Ther. Adv. Med. Oncol., 2018, 10, 1–18 Search PubMed.
- R. Scully, A. Panday, R. Elango and N. A. Willis, Nat. Rev. Mol. Cell Biol., 2011, 20, 698–714 CrossRef.
- N. J. O’Neil, M. L. Bailey and P. Hieter, Nat. Rev. Genet., 2017, 18, 613–623 CrossRef.
- P. G. Pilié, C. Tang, G. B. Mills and T. A. Yap, Nat. Rev. Clin. Oncol., 2019, 16, 81–104 CrossRef.
- D. Varshney, J. Spiegel, K. Zyner, D. Tannahill and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2020, 21, 459–474 CrossRef CAS.
- R. Hänsel-Hertsch, M. Di Antonio and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2017, 18, 279–284 CrossRef.
- A. N. Khristich and S. M. Mirkin, J. Biol. Chem., 2020, 295, 4134–4170 CrossRef CAS.
- C. K. Kwok and C. J. Merrick, Trends Biotechnol., 2017, 35, 997–1013 CrossRef CAS.
- S. Neidle, J. Med. Chem., 2016, 59, 5987–6011 CrossRef CAS.
- S. Neidle, Nat. Rev. Chem., 2017, 1, 0041 CrossRef CAS.
- G. A. Holdgate, T. D. Meek and R. L. Grimley, Nat. Rev. Drug Discovery, 2018, 17, 115–132 CrossRef CAS.
- N. Puget, K. M. Miller and G. Legube, DNA Repair, 2019, 81, 102661 CrossRef CAS.
- J. Zhao, A. Bacolla, G. Wang and K. M. Vasquez, Cell. Mol. Life Sci., 2010, 67, 43–62 CrossRef CAS.
- B. P. Belotserkovskii, S. M. Mirkin and P. C. Hanawalt, Chem. Rev., 2013, 113, 8620–8637 CrossRef CAS.
- R. Dahm, Dev. Biol., 2005, 278, 274–288 CrossRef CAS.
- A. Hollaender and B. M. Duggar, J. Bacteriol., 1938, 36, 17–37 CrossRef CAS.
- E. C. Friedberg, DNA Repair, 2015, 33, 35–42 CrossRef CAS.
- E. C. Friedberg, Cell Res., 2008, 18, 3–7 CrossRef CAS.
- A. Kelner, Proc. Natl. Acad. Sci. U. S. A., 1949, 34, 73–79 CrossRef.
- R. Dulbecco, Nature, 1949, 163, 949–950 CrossRef CAS.
- G. B. Sancar, Mutat. Res., 2000, 451, 25–37 CrossRef CAS.
- J. D. Watson and F. Crick, Nature, 1953, 171, 737–738 CrossRef CAS.
- C. Dennis and P. Campbell, Nature, 1953, 171, 740–741 CrossRef.
- M. H. F. Wilkins, A. R. Wilson and H. R. Wilson, Nature, 1953, 171, 738–740 CrossRef CAS.
- R. Beukers, J. Ylstra and W. Berends, Recl. Trav. Chim. Pays-Bas, 1958, 77, 729–732 CrossRef CAS.
- S. Minato and H. Werbin, Photochem. Photobiol., 1972, 15, 97–100 CrossRef CAS.
- J. J. Madden and H. Werbin, Biochemistry, 1974, 13, 2149–2154 CrossRef CAS.
- T. Lindahl and D. E. Barnes, Cold Spring Harbor Symp. Quant. Biol., 2000, 65, 127–133 CrossRef CAS.
- A. Ciccia and S. J. Elledge, Mol. Cell, 2010, 40, 179–204 CrossRef CAS.
- D. Cortez, DNA Repair, 2015, 32, 149–157 CrossRef CAS.
- H. Técher, S. Koundrioukoff, A. Nicolas and M. Debatisse, Nat. Rev. Genet., 2017, 18, 535–550 CrossRef.
- T. Helleday, S. Eshtad and S. Nik-zainal, Nat. Rev. Genet., 2014, 15, 585–598 CrossRef CAS.
- A. N. Blackford and S. P. Jackson, Mol. Cell, 2017, 66, 801–817 CrossRef CAS.
- S. I. Hajdu, Cancer, 2011, 117, 1097–1102 CrossRef.
- S. I. Hajdu, Cancer, 2012, 118, 4914–4928 CrossRef.
- D. Vilches, G. Alburquerque and R. Ramirez-Tagle, Educ. Quim., 2016, 27, 233–236 Search PubMed.
- J. Hirsch, J. Am. Med. Assoc., 2006, 296, 1518–1520 CrossRef CAS.
- K. W. Kohn, Cancer Res., 1996, 56, 5533–5546 CAS.
- L. H. Hurley, Nat. Rev. Cancer, 2002, 2, 188–200 CrossRef CAS.
- A. Gilman and F. S. Philips, Science, 1946, 103, 409–436 CrossRef CAS.
- C. P. Rhoads, Ca-Cancer J. Clin., 1978, 28, 306–312 CrossRef CAS.
- P. Christakis, Yale J. Biol. Med., 2011, 84, 169–172 Search PubMed.
- L. Goodman, M. Wintrobe, W. Dameshek, M. Goodman, A. Gilman and M. McLennan, J. Am. Med. Assoc., 1946, 132, 126–132 CrossRef CAS.
- V. T. DeVita and E. Chu, Cancer Res., 2008, 68, 8643–8653 CrossRef CAS.
- K. W. Kohn, Cancer Res., 1977, 37, 1450–1454 CAS.
- S. Mukherjee, The Emperor of all Maladies: A Biography of Cancer, Scribner, New York, Toronto, 1st edn, 2010 Search PubMed.
- S. Farber, L. K. Diamond, R. D. Mercer, R. F. Sylvester and J. A. Wolff, N. Engl. J. Med., 1948, 238, 787–793 CrossRef CAS.
- C. Heidelberger, N. K. Chaudhuri, P. Danneberg, D. Mooren, L. Griesbach, R. Duschinsky, R. J. Schnitzer, E. Pleven and J. Scheiner, Nature, 1957, 179, 663–666 CrossRef CAS.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS.
- B. Druker, S. Tamura, E. Buchdunger, S. Ohno, G. M. Segal, S. Fanning, J. Zimmermann and N. B. Lydon, Nat. Med., 1996, 2, 561–566 CrossRef CAS.
- T. Hunter, J. Clin. Invest., 2007, 117, 2036–2043 CrossRef CAS.
- Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS.
- K. Chválová, V. Brabec and J. Kašpárková, Nucleic Acids Res., 2007, 35, 1812–1821 CrossRef.
- J. L. Delgado, C. M. Hsieh, N. L. Chan and H. Hiasa, Biochem. J., 2018, 475, 373–398 CrossRef CAS.
- J. Marinello, M. Delcuratolo and G. Capranico, Int. J. Mol. Sci., 2018, 19, 3480 CrossRef.
- J. L. Nitiss, Nat. Rev. Cancer, 2009, 9, 338–350 CrossRef CAS.
- J. Stingele, R. Bellelli and S. J. Boulton, Nat. Rev. Mol. Cell Biol., 2017, 18, 563–573 CrossRef CAS.
- Y. Pommier, Nat. Rev. Cancer, 2006, 6, 789–802 CrossRef CAS.
- G. L. Patrick, An Introduction to Medicinal Chemistry, Oxford University Press, Oxford, UK, 5th edn, 2013 Search PubMed.
- R. A. Burrell, N. McGranahan, J. Bartek and C. Swanton, Nature, 2013, 501, 338–345 CrossRef CAS.
- J. Huszno, Z. Kołosza and E. W. A. Grzybowska, Oncol. Lett., 2019, 17, 1986–1995 CAS.
- I. Gorodetska, I. Kozeretska and A. Dubrovska, J. Cancer, 2019, 10, 2109–2127 CrossRef CAS.
- S. Negrini, V. G. Gorgoulis and T. D. Halazonetis, Nat. Rev. Mol. Cell Biol., 2010, 11, 220–228 CrossRef CAS.
- E. R. Kastenhuber and S. W. Lowe, Cell, 2017, 170, 1062–1078 CrossRef CAS.
- L. H. Pearl, A. C. Schierz, S. E. Ward, B. Al-Lazikani and F. M. G. Pearl, Nat. Rev. Cancer, 2015, 15, 166–180 CrossRef CAS.
- P. C. Hanawalt, DNA Repair, 2015, 36, 2–7 CrossRef CAS.
- Y. Shiloh and Y. Ziv, Nat. Rev. Mol. Cell Biol., 2013, 14, 197–210 CrossRef CAS.
- Y. Shiloh, Nat. Rev. Cancer, 2003, 3, 155–168 CrossRef CAS.
- J. Stavnezer and C. E. Schrader, J. Immunol., 2014, 193, 5370–5378 CrossRef CAS.
- K. J. Zanotti and P. J. Gearhart, DNA Repair, 2016, 38, 110–116 CrossRef CAS.
- I. Lam and S. Keeney, Cold Spring Harbor Perspect. Biol., 2015, 7, 0016634 Search PubMed.
- T. Lindahl, Angew. Chem., Int. Ed., 2016, 55, 8528–8534 CrossRef CAS.
- P. Modrich, Angew. Chem., Int. Ed., 2016, 55, 8490–8501 CrossRef CAS.
- A. Sancar, Angew. Chem., Int. Ed., 2016, 55, 8502–8527 CrossRef CAS.
- M. Olivieri, T. Cho, A. Álvarez-Quilón, K. Li, M. J. Schellenberg, M. Zimmermann, N. Hustedt, S. E. Rossi, S. Adam, H. Melo, A. M. Heijink, G. Sastre-Moreno, N. Moatti, R. Szilard, A. McEwan, A. K. Ling, A. Serrano-Benitez, T. Ubhi, S. Feng, J. Pawling, I. Delgado-Sainz, M. W. Ferguson, J. W. Ddennis, G. W. Brown, F. Cortés-Ledesma, R. S. Williams, A. Martin, D. Xu and D. Durocher, Cell, 2020, 182, 481–496 CrossRef CAS.
- R. Abbotts and D. M. Wilson, Free Radical Biol. Med., 2017, 107, 228–244 CrossRef CAS.
- J. Jiricny, Nat. Rev. Mol. Cell Biol., 2006, 7, 335–346 CrossRef CAS.
- G. X. Reyes, T. T. Schmidt, R. D. Kolodner and H. Hombauer, Chromosoma, 2015, 124, 443–462 CrossRef CAS.
- A. Sancar, Nat. Genet., 1999, 21, 247–249 CrossRef CAS.
- T. Nakano, S. Morishita, A. Katafuchi, M. Matsubara, Y. Horikawa, H. Terato, A. M. H. Salem, S. Izumi, S. P. Pack, K. Makino and H. Ide, Mol. Cell, 2007, 28, 147–158 CrossRef CAS.
- D. J. Baker, G. Wuenschell, L. Xia, J. Termini, S. E. Bates, A. D. Riggs and T. R. O’Connor, J. Biol. Chem., 2007, 282, 22592–22604 CrossRef CAS.
- G. Spivak and A. K. Ganesan, DNA Repair, 2014, 19, 64–70 CrossRef CAS.
- P. M. J. Burgers and T. A. Kunkel, Annu. Rev. Biochem., 2017, 86, 417–438 CrossRef CAS.
- L. J. Martin, J. Neuropathol. Exp. Neurol., 2008, 67, 377–387 CrossRef CAS.
- B. Sedgwick, P. A. Bates, J. Paik, S. C. Jacobs and T. Lindahl, DNA Repair, 2007, 6, 429–442 CrossRef CAS.
- L. C. Huang, K. C. Clarkin and G. M. Wahl, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4827–4832 CrossRef CAS.
- A. Pipier, M. Bossaert, J. F. Riou, C. Noirot, L.-T. Nguyên, R.-F. Serre, O. Bouchez, E. Defrancq, P. Calsou, S. Britton and D. Gomez, bioRxiv, 2020 DOI:10.1101/2020.02.18.953851.
- R. Rodriguez, K. M. Miller, J. V. Forment, C. R. Bradshaw, M. Nikan, S. Britton, T. Oelschlaegel, B. Xhemalce, S. Balasubramanian and S. P. Jackson, Nat. Chem. Biol., 2012, 8, 301–310 CrossRef CAS.
- K. Karanam, R. Kafri, A. Loewer and G. Lahav, Mol. Cell, 2012, 47, 320–329 CrossRef CAS.
- L. Deriano and D. B. Roth, Annu. Rev. Genet., 2013, 47, 433–455 CrossRef CAS.
- A. Shibata, P. Jeggo and M. Löbrich, DNA Repair, 2018, 71, 164–171 CrossRef CAS.
- R. L. Flynn and L. Zou, Crit. Rev. Biochem. Mol. Biol., 2010, 45, 266–275 CrossRef CAS.
- W. D. Wright, S. S. Shah and W. D. Heyer, J. Biol. Chem., 2018, 293, 10524–10535 CrossRef CAS.
- W. Zhao, J. B. Steinfeld, F. Liang, X. Chen, D. G. Maranon, C. Jian Ma, Y. Kwon, T. Rao, W. Wang, C. Sheng, X. Song, Y. Deng, J. Jimenez-Sainz, L. Lu, R. B. Jensen, Y. Xiong, G. M. Kupfer, C. Wiese, E. C. Greene and P. Sung, Nature, 2017, 550, 360–365 CrossRef CAS.
- A. Datta and R. M. Brosh, Genes, 2019, 10, 170 CrossRef CAS.
- G.-L. Moldovan and A. D. D’Andrea, Annu. Rev. Genet., 2009, 43, 223–249 CrossRef CAS.
- A. M. R. Taylor, C. Rothblum-Oviatt, N. A. Ellis, I. D. Hickson, S. Meyer, T. O. Crawford, A. Smogorzewska, B. Pietrucha, C. Weemaes and G. S. Stewart, Nat. Rev. Dis. Prim., 2019, 5, 64 CrossRef.
- J. Niraj, A. Färkkilä and A. D. D’Andrea, Annu. Rev. Cancer Biol., 2019, 3, 457–478 CrossRef.
- M. C. Kottemann and A. Smogorzewska, Nature, 2013, 493, 356–363 CrossRef CAS.
- P. Tonzi, Y. Yin, C. W. T. Lee, E. Rothenberg and T. T. Huang, eLife, 2018, 7, e41426 CrossRef.
- H. L. Williams, M. E. Gottesman and J. Gautier, Mol. Cell, 2012, 47, 140–147 CrossRef CAS.
- J. Murai, S. Y. N. Huang, B. B. Das, A. Renaud, Y. Zhang, J. H. Doroshow, J. Ji, S. Takeda and Y. Pommier, Cancer Res., 2012, 72, 5588–5599 CrossRef CAS.
- K. W. Caldecott, Nat. Rev. Genet., 2008, 9, 619–631 CrossRef CAS.
- C. J. Lord and A. Ashworth, Science, 2017, 355, 1152–1158 CrossRef CAS.
- L. Palazzo and I. Ahel, Biochem. Soc. Trans., 2018, 46, 1681–1695 CrossRef CAS.
- B. A. Gibson, Y. Zhang, H. Jiang, K. M. Hussey, J. H. Shrimp, H. Lin, F. Schwede, Y. Yu and W. L. Kraus, Science, 2016, 353, 45–50 CrossRef CAS.
- C. Beck, I. Robert, B. Reina-San-Martin, V. Schreiber and F. Dantzer, Exp. Cell Res., 2014, 329, 18–25 CrossRef CAS.
- S. L. Rulten, A. E. O. Fisher, I. Robert, M. C. Zuma, M. Rouleau, L. Ju, G. Poirier, B. Reina-San-Martin and K. W. Caldecott, Mol. Cell, 2011, 41, 33–45 CrossRef CAS.
- C. Beck, C. Boehler, J. Guirouilh Barbat, M. E. Bonnet, G. Illuzzi, P. Ronde, L. R. Gauthier, N. Magroun, A. Rajendran, B. S. Lopez, R. Scully, F. D. Boussin, V. Schreiber and F. Dantzer, Nucleic Acids Res., 2014, 42, 5616–5632 CrossRef CAS.
- T. A. Day, J. V. Layer, J. Patrick Cleary, S. Guha, K. E. Stevenson, T. Tivey, S. Kim, A. C. Schinzel, F. Izzo, J. Doench, D. E. Root, W. C. Hahn, B. D. Price and D. M. Weinstock, Nat. Commun., 2017, 8, 15110 CrossRef.
- N. Hustedt and D. Durocher, Nat. Cell Biol., 2017, 19, 1–9 CrossRef CAS.
- J. Falck, J. Coates and S. P. Jackson, Nature, 2005, 434, 605–611 CrossRef CAS.
- A. Syed and J. A. Tainer, Annu. Rev. Biochem., 2018, 87, 263–294 CrossRef CAS.
- S. Britton, J. Coates and S. P. Jackson, J. Cell Biol., 2013, 202, 579–595 CrossRef CAS.
- M. Stucki, J. A. Clapperton, D. Mohammad, M. B. Yaffe, S. J. Smerdon and S. P. Jackson, Cell, 2005, 123, 1213–1226 CrossRef CAS.
- M. S. Lee, R. A. Edwards, G. L. Thede and J. N. M. Glover, J. Biol. Chem., 2005, 280, 32053–32056 CrossRef CAS.
- M. Goldberg, M. Stucki, J. Falck, D. D’Amours, D. Rahman, D. Pappin, J. Bartek and S. P. Jackson, Nature, 2003, 421, 952–956 CrossRef CAS.
- R. Scully and A. Xie, Mutat. Res., 2013, 750, 5–14 CrossRef CAS.
- J. R. Chapman and S. P. Jackson, EMBO Rep., 2008, 9, 795–801 CrossRef CAS.
- F. J. Hari, C. Spycher, S. Jungmichel, L. Pavic and M. Stucki, EMBO Rep., 2010, 11, 387–392 CrossRef CAS.
- S. Burma, B. P. Chen, M. Murphy, A. Kurimasa and D. J. Chen, J. Biol. Chem., 2001, 276, 42462–42467 CrossRef CAS.
- J. D. Friesner, B. Liu, K. Culligan and A. B. Britt, Mol. Biol. Cell, 2005, 16, 2566–2576 CrossRef CAS.
- P. Caron, F. Aymard, J. S. Iacovoni, S. Briois, Y. Canitrot, B. Bugler, L. Massip, A. Losada and G. Legube, PLoS Genet., 2012, 8, e1002460 CrossRef CAS.
- I. Litwin, E. Pilarczyk and R. Wysocki, Genes, 2018, 9, 581 CrossRef.
- C. Meisenberg, S. I. Pinder, S. R. Hopkins, S. K. Wooller, G. Benstead-Hume, F. M. G. Pearl, P. A. Jeggo and J. A. Downs, Mol. Cell, 2019, 73, 212–223 CrossRef CAS.
- Z. Lou, K. Minter-Dykhouse, X. Wu and J. Chen, Nature, 2003, 421, 957–961 CrossRef CAS.
- G. S. Stewart, B. Wang, C. R. Bigneli, A. M. R. Taylor and S. J. Elledge, Nature, 2003, 421, 961–966 CrossRef CAS.
- W. M. Bonner, C. E. Redon, J. S. Dickey, A. J. Nakamura, O. A. Sedelnikova, S. Solier and Y. Pommier, Nat. Rev. Cancer, 2008, 8, 957–967 CrossRef CAS.
- P. Chanut, S. Britton, J. Coates, S. P. Jackson and P. Calsou, Nat. Commun., 2016, 7, 12889 CrossRef.
- B. P. C. Chen, N. Uematsu, J. Kobayashi, Y. Lerenthal, A. Krempler, H. Yajima, M. Löbrich, Y. Shiloh and D. J. Chen, J. Biol. Chem., 2007, 282, 6582–6587 CrossRef CAS.
- D. Normanno, A. Négrel, A. J. de Melo, S. Betzi, K. Meek and M. Modesti, eLife, 2017, 6, e22900 CrossRef.
- M. Hammel, Y. Yu, B. L. Mahaney, B. Cai, R. Ye, B. M. Phipps, R. P. Rambo, G. L. Hura, M. Pelikan, S. So, R. M. Abolfath, D. J. Chen, S. P. Lees-Miller and J. A. Tainer, J. Biol. Chem., 2010, 285, 1414–1423 CrossRef CAS.
- S. Britton, P. Chanut, C. Delteil, N. Barboule, P. Frit and P. Calsou, Nucleic Acids Res., 2020 DOI:10.1093/nar/gkaa723.
- M. D. Wilson, S. Benlekbir, A. Fradet-Turcotte, A. Sherker, J. P. Julien, A. McEwan, S. M. Noordermeer, F. Sicheri, J. L. Rubinstein and D. Durocher, Nature, 2016, 536, 100–103 CrossRef CAS.
- S. M. Noordermeer, S. Adam, D. Setiaputra, M. Barazas, S. J. Pettitt, A. K. Ling, M. Olivieri, A. Álvarez-Quilón, N. Moatti, M. Zimmermann, S. Annunziato, D. B. Krastev, F. Song, I. Brandsma, J. Frankum, R. Brough, A. Sherker, S. Landry, R. K. Szilard, M. M. Munro, A. McEwan, T. G. de Rugy, Z. Y. Lin, T. Hart, J. Moffat, A. C. Gingras, A. Martin, H. van Attikum, J. Jonkers, C. J. Lord, S. Rottenberg and D. Durocher, Nature, 2018, 560, 117–121 CrossRef CAS.
- S. Panier and S. J. Boulton, Nat. Rev. Mol. Cell Biol., 2014, 15, 7–18 CrossRef CAS.
- K. A. Cimprich and D. Cortez, Nat. Rev. Mol. Cell Biol., 2008, 9, 616–627 CrossRef CAS.
- V. Costanzo, D. Shechter, P. J. Lupardus, K. A. Cimprich, M. Gottesman and J. Gautier, Mol. Cell, 2003, 11, 203–213 CrossRef CAS.
- F. A. Derheimer, H. M. O’Hagan, H. M. Krueger, S. Hanasoge, M. T. Paulsen and M. Ljungman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12778–12783 CrossRef CAS.
- K. Trenz, E. Smith, S. Smith and V. Costanzo, EMBO J., 2006, 25, 1764–1774 CrossRef CAS.
- J. M. Merchut-Maya, J. Bartek and A. Maya-Mendoza, DNA Repair, 2019, 81, 102654 CrossRef CAS.
- L. I. Toledo, M. Altmeyer, M. B. Rask, C. Lukas, D. H. Larsen, L. K. Povlsen, S. Bekker-Jensen, N. Mailand, J. Bartek and J. Lukas, Cell, 2013, 155, 1088 CrossRef CAS.
- E. J. Brown and D. Baltimore, Genes Dev., 2000, 14, 397–402 CAS.
- A. De Klein, M. Muijtjens, R. Van Os, Y. Verhoeven, B. Smit, A. M. Carr, A. R. Lehmann and J. H. J. Hoeijmakers, Curr. Biol., 2000, 10, 479–482 CrossRef CAS.
- A. Jazayeri, J. Falck, C. Lukas, J. Bartek, G. C. M. Smith, J. Lukas and S. P. Jackson, Nat. Cell Biol., 2006, 8, 37–45 CrossRef CAS.
- M. Malumbres and M. Barbacid, Nat. Rev. Cancer, 2009, 9, 153–166 CrossRef CAS.
- A. B. Williams and B. Schumacher, Cold Spring Harbor Perspect. Med., 2016, 6, a026070 CrossRef.
- K. J. Barnum and M. J. O’Connell, Methods Mol. Biol., 2014, 1170, 29–40 CrossRef.
- A. M. Abukhdeir and B. H. Park, Expert Rev. Mol. Med., 2008, 10, e19 CrossRef.
- P. Nurse, Nature, 2005, 432, 557 CrossRef.
- C. J. Matheson, D. S. Backos and P. Reigan, Trends Pharmacol. Sci., 2016, 37, 872–881 CrossRef CAS.
- M. Chen, C. E. Ryan and H. Piwnica-Worms, Mol. Cell. Biol., 2003, 23, 7488–7497 CrossRef CAS.
- H. Löffler, B. Rebacz, A. D. Ho, J. Lukas, J. Bartek and A. Krämer, Cell Cycle, 2006, 5, 2543–2547 CrossRef.
- M. Donzelli and G. F. Draetta, EMBO Rep., 2003, 4, 671–677 CrossRef CAS.
- H. Wang, X. Zhang, L. Teng and R. J. Legerski, Exp. Cell Res., 2015, 334, 350–358 CrossRef.
- T. A. Yap, R. Plummer, N. S. Azad and T. Helleday, Am. Soc. Clin. Oncol. Educ. B, 2019, 185–195 CrossRef.
- M. Dobbelstein and C. S. Sørensen, Nat. Rev. Drug Discovery, 2015, 14, 405–423 CrossRef CAS.
- S. Murata, C. Zhang, N. Finch, K. Zhang, L. Campo and E. K. Breuer, BioMed Res. Int., 2016, 2346585 Search PubMed.
- W. G. Kaelin, Nat. Rev. Cancer, 2005, 5, 689–698 CrossRef CAS.
- M. Shaheen, C. Allen, J. A. Nickoloff and R. Hromas, Blood, 2011, 117, 6074–6082 CrossRef CAS.
- T. Dobzhansky, Genetics, 1946, 31, 269–290 CAS.
- M. Keung, Y. Wu and J. Vadgama, J. Clin. Med., 2019, 8, 435 CrossRef CAS.
- J. S. Brown, B. O’Carrigan, S. P. Jackson and T. A. Yap, Cancer Discovery, 2017, 7, 20–37 CrossRef CAS.
- P. Munster, M. Mita, A. Mahipal, J. Nemunaitis, C. Massard, T. Mikkelsen, C. Cruz, L. Paz-Ares, M. Hidalgo, D. Rathkopf, G. Blumenschein, D. C. Smith, B. Eichhorst, T. Cloughesy, E. H. Filvaroff, S. Li, H. Raymon, H. de Haan, K. Hege and J. C. Bendell, Cancer Manage. Res., 2019, 11, 10463–10476 CrossRef CAS.
- B. Van Triest, L. Damstrup, J. Falkenius, V. Budach, E. Troost, M. Samuels, T. Goddemeier and P. F. Geertsen, J. Clin. Oncol., 2017, 35, e14048 Search PubMed.
- B. Haynes, J. Murai and J. M. Lee, Cancer Treat. Rev., 2018, 71, 1–7 CrossRef CAS.
- T. P. Matthews, A. M. Jones and I. Collins, Expert Opin. Drug Discovery, 2013, 8, 621–640 CrossRef CAS.
- J. Shendure, S. Balasubramanian, G. M. Church, W. Gilbert, J. Rogers, J. A. Schloss and R. H. Waterston, Nature, 2017, 550, 345–353 CrossRef CAS.
- C. Sánchez-Martínez, M. J. Lallena, S. G. Sanfeliciano and A. de Dios, Bioorg. Med. Chem. Lett., 2019, 29, 126637 CrossRef.
- A. Bradbury, S. Hall, N. Curtin and Y. Drew, Pharmacol. Ther., 2020, 207, 107450 CrossRef CAS.
- S. Rundle, A. Bradbury, Y. Drew and N. J. Curtin, Cancers, 2017, 9, 41 CrossRef.
- Y. Wang and D. J. Patel, Structure, 1993, 1, 263–282 CrossRef CAS.
- K. N. Luu, A. T. Phan, V. Kuryavyi, L. Lacroix and D. J. Patel, J. Am. Chem. Soc., 2006, 128, 9963–9970 CrossRef CAS.
- G. N. Parkinson, M. P. H. Lee and S. Neidle, Nature, 2002, 417, 876–880 CrossRef CAS.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef CAS.
- D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627–8637 CrossRef CAS.
- L. Stefan and D. Monchaud, Nat. Rev. Chem., 2019, 3, 650–668 CrossRef.
- M. Gellert, M. N. Lipsett and D. R. Davies, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 2013–2018 CrossRef CAS.
- V. S. Chambers, G. Marsico, J. M. Boutell, M. Di Antonio, G. P. Smith and S. Balasubramanian, Nat. Biotechnol., 2015, 33, 877–881 CrossRef.
- R. Hänsel-Hertsch, D. Beraldi, S. V. Lensing, G. Marsico, K. Zyner, A. Parry, M. Di Antonio, J. Pike, H. Kimura, M. Narita, D. Tannahill and S. Balasubramanian, Nat. Genet., 2016, 48, 1267–1272 CrossRef.
- M. Di Antonio, A. Ponjavic, A. Radzevičius, R. T. Ranasinghe, M. Catalano, X. Zhang, J. Shen, L. M. Needham, S. F. Lee, D. Klenerman and S. Balasubramanian, Nat. Chem., 2020, 12, 832–837 CrossRef CAS.
- J. Spiegel, S. Adhikari and S. Balasubramanian, Trends Chem., 2020, 2, 123–136 CrossRef CAS.
- A. K. Todd, M. Johnston and S. Neidle, Nucleic Acids Res., 2005, 33, 2901–2907 CrossRef CAS.
- N. Maizels, EMBO Rep., 2015, 16, 910–922 CrossRef CAS.
- G. Marsico, V. S. Chambers, A. B. Sahakyan, P. McCauley, J. M. Boutell, M. Di Antonio and S. Balasubramanian, Nucleic Acids Res., 2019, 47, 3862–3874 CrossRef CAS.
- N. Kim, Curr. Med. Chem., 2017, 26, 2898–2917 CrossRef.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS.
- T. M. Bryan, Molecules, 2019, 24, 3439 CrossRef CAS.
- C. Cayrou, B. Ballester, I. Peiffer, R. Fenouil, P. Coulombe, J. C. Andrau, J. Van Helden and M. Méchali, Genome Res., 2015, 25, 1873–1885 CrossRef CAS.
- P. Prorok, M. Artufel, A. Aze, P. Coulombe, I. Peiffer, L. Lacroix, A. Guédin, J. L. Mergny, J. Damaschke, A. Schepers, B. Ballester and M. Méchali, Nat. Commun., 2019, 10, 3274 CrossRef CAS.
- Y. Xu, Chem. Soc. Rev., 2011, 40, 2719–2740 RSC.
- K. Turner, V. Vasu and D. Griffin, Cells, 2019, 8, 73 CrossRef CAS.
- J. D. Watson, Nature, New Biol., 1972, 239, 197–201 CrossRef CAS.
- A. M. Olovnikov, J. Theor. Biol., 1973, 41, 181–190 CrossRef CAS.
- P. Martínez and M. A. Blasco, J. Cell Biol., 2017, 216, 875–887 CrossRef.
- C. López-Otín, M. A. Blasco, L. Partridge, M. Serrano and G. Kroemer, Cell, 2013, 153, 1194 CrossRef.
- A. De Cian, L. Lacroix, C. Douarre, N. Temime-Smaali, C. Trentesaux, J. F. Riou and J. L. Mergny, Biochimie, 2008, 90, 131–155 CrossRef CAS.
- H. Q. Yu, D. Miyoshi and N. Sugimoto, J. Am. Chem. Soc., 2006, 128, 15461–15468 CrossRef CAS.
- J. S. Smith, Q. Chen, L. A. Yatsunyk, J. M. Nicoludis, M. S. Garcia, R. Kranaster, S. Balasubramanian, D. Monchaud, M. P. Teulade-Fichou, L. Abramowitz, D. C. Schultz and F. B. Johnson, Nat. Struct. Mol. Biol., 2011, 18, 478–486 CrossRef CAS.
- E. H. Blackburn, Angew. Chem., Int. Ed., 2010, 49, 7405–7421 CrossRef CAS.
- C. W. Greider, Angew. Chem., Int. Ed., 2010, 49, 7422–7439 CrossRef CAS.
- J. W. Szostak, Angew. Chem., Int. Ed., 2010, 49, 7386–7404 CrossRef CAS.
- E. H. Blackburn, C. W. Greider and J. W. Szostak, Nat. Med., 2006, 12, 1133–1138 CrossRef CAS.
- Y. Deng, S. S. Chan and S. Chang, Nat. Rev. Cancer, 2008, 8, 450–458 CrossRef CAS.
- O. Mendoza, A. Bourdoncle, J. B. Boulé, R. M. Brosh and J. L. Mergny, Nucleic Acids Res., 2016, 44, 1989–2006 CrossRef CAS.
- M. Sauer and K. Paeschke, Biochem. Soc. Trans., 2017, 45, 1173–1182 CrossRef CAS.
- R. M. Brosh, Nat. Rev. Cancer, 2013, 13, 542–558 CrossRef CAS.
- R. M. Brosh and S. W. Matson, Genes, 2020, 11, 255 CrossRef.
- K. N. Estep and R. M. Brosh, Biochem. Soc. Trans., 2017, 46, 77–95 CrossRef.
- P. Lansdorp and N. van Wietmarschen, Genes, 2019, 10, 870 CrossRef CAS.
- L. K. Lerner and J. E. Sale, Genes, 2019, 10, 95 CrossRef CAS.
- M. E. Fairman-Williams, U. P. Guenther and E. Jankowsky, Curr. Opin. Struct. Biol., 2010, 20, 313–324 CrossRef CAS.
- M. R. Singleton, M. S. Dillingham and D. B. Wigley, Annu. Rev. Biochem., 2007, 76, 23–50 CrossRef CAS.
- H. Sun, J. K. Karow, I. D. Hickson and N. Maizels, J. Biol. Chem., 1998, 273, 27587–27592 CrossRef CAS.
- P. Mohaghegh, J. K. Karow, R. M. Brosh, V. A. Bohr and I. D. Hickson, Nucleic Acids Res., 2001, 29, 2843–2849 CrossRef CAS.
- J. L. Li, R. J. Harrison, A. P. Reszka, R. M. Brosh, V. A. Bohr, S. Neidle and I. D. Hickson, Biochemistry, 2001, 40, 15194–15202 CrossRef CAS.
- H. Han, R. J. Bennett and L. H. Hurley, Biochemistry, 2000, 39, 9311–9316 CrossRef CAS.
- H. Sun, R. J. Bennett and N. Maizels, Nucleic Acids Res., 1999, 27, 1978–1984 CrossRef CAS.
- M. Fry and L. A. Loeb, J. Biol. Chem., 1999, 274, 12797–12802 CrossRef CAS.
- X. Wu and N. Maizels, Nucleic Acids Res., 2001, 29, 1765–1771 CrossRef CAS.
- S. D. Creacy, E. D. Routh, F. Iwamoto, Y. Nagamine, S. A. Akman and J. P. Vaughn, J. Biol. Chem., 2008, 283, 34626–34634 CrossRef CAS.
- J. P. Vaughn, S. D. Creacy, E. D. Routh, C. Joyner-Butt, G. S. Jenkins, S. Pauli, Y. Nagamine and S. A. Akman, J. Biol. Chem., 2005, 280, 38117–38120 CrossRef CAS.
- M. C. Chen, P. Murat, K. Abecassis, A. R. Ferre-D’Amare and S. Balasubramanian, Nucleic Acids Res., 2015, 43, 2223–2231 CrossRef CAS.
- C. Ribeyre, J. Lopes, J. B. Boulé, A. Piazza, A. Guédin, V. A. Zakian, J. L. Mergny and A. Nicolas, PLoS Genet., 2009, 5, e1000475 CrossRef.
- A. Piazza, J. B. Boulé, J. Lopes, K. Mingo, E. Largy, M. P. Teulade-Fichou and A. Nicolas, Nucleic Acids Res., 2010, 38, 4337–4348 CrossRef CAS.
- K. Paeschke, M. L. Bochman, P. Daniela Garcia, P. Cejka, K. L. Friedman, S. C. Kowalczykowski and V. A. Zakian, Nature, 2013, 497, 458–462 CrossRef CAS.
- S. Dehghani-Tafti, V. Levdikov, A. A. Antson, B. Bax and C. M. Sanders, Nucleic Acids Res., 2019, 47, 3208–3222 CrossRef CAS.
- J. B. Vannier, S. Sandhu, M. I. R. Petalcorin, X. Wu, Z. Nabi, H. Ding and S. J. Boulton, Science, 2013, 342, 239–242 CrossRef CAS.
- J. B. Vannier, V. Pavicic-Kaltenbrunner, M. I. R. Petalcorin, H. Ding and S. J. Boulton, Cell, 2012, 149, 795–806 CrossRef CAS.
- T. B. C. London, L. J. Barber, G. Mosedale, G. P. Kelly, S. Balasubramanian, I. D. Hickson, S. J. Boulton and K. Hiom, J. Biol. Chem., 2008, 283, 36132–36139 CrossRef CAS.
- Y. Wu, K. Shin-ya and R. M. Brosh, Mol. Cell. Biol., 2008, 28, 4116–4128 CrossRef CAS.
- S. K. Bharti, J. A. Sommers, F. George, J. Kuper, F. Hamon, K. Shin-Ya, M. P. Teulade-Fichou, C. Kisker and R. M. Brosh, J. Biol. Chem., 2013, 288, 28217–28229 CrossRef CAS.
- Y. Wu, J. A. Sommers, I. Khan, J. P. De Winter and R. M. Brosh, J. Biol. Chem., 2012, 287, 1007–1021 CrossRef CAS.
- L. T. Gray, A. C. Vallur, J. Eddy and N. Maizels, Nat. Chem. Biol., 2014, 10, 313–318 CrossRef CAS.
- A. R. Lehmann, Genes Dev., 2001, 15, 15–23 CrossRef CAS.
- W. C. Drosopoulos, S. T. Kosiyatrakul and C. L. Schildkraut, J. Cell Biol., 2015, 210, 191–208 CrossRef CAS.
- N. Van Wietmarschen, S. Merzouk, N. Halsema, D. C. J. Spierings, V. Guryev and P. Lansdorp, Nat. Commun., 2018, 9, 271 CrossRef.
- T. De Lange, Genes Dev., 2005, 19, 2100–2110 CrossRef CAS.
- I. Cheung, M. Schertzer, A. Rose and P. Lansdorp, Nat. Genet., 2002, 31, 405–409 CrossRef CAS.
- R. A. Schwab, J. Nieminuszczy, K. Shin-ya and W. Niedzwiedz, J. Cell Biol., 2013, 201, 33–48 CrossRef CAS.
- K. Paeschke, J. A. Capra and V. A. Zakian, Cell, 2011, 145, 678–691 CrossRef CAS.
- A. Piazza, M. Adrian, F. Samazan, B. Heddi, F. Hamon, A. Serero, J. Lopes, M. Teulade-Fichou, A. T. Phan and A. Nicolas, EMBO J., 2015, 34, 1718–1734 CrossRef CAS.
- L. Maestroni, J. Audry, P. Luciano, S. Coulon, V. Géli and Y. Corda, Cell Stress, 2020, 4, 48–63 CrossRef CAS.
- S. Jimeno, R. Camarillo, F. Mejías-Navarro, M. J. Fernández-Ávila, I. Soria-Bretones, R. Prados-Carvajal and P. Huertas, Cell Rep., 2018, 24, 3262–3273 CrossRef CAS.
- J. F. Moruno-Manchon, P. Lejault, Y. Wang, B. McCauley, P. Honarpisheh, D. A. Morales Scheihing, S. Singh, W. Dang, N. Kim, A. Urayama, L. Zhu, D. Monchaud, L. D. McCullough and A. S. Tsvetkov, eLife, 2020, 9, e52283 CrossRef.
- J. Tan, X. Wang, L. Phoon, H. Yang and L. Lan, FEBS Lett., 2020, 594, 1359–1367 CrossRef CAS.
- G. Beadle and L. Pauling, Eng. Sci., 1954, 17, 9 Search PubMed.
- K. H. Altmann, J. Buchner, H. Kessler, F. Diederich, B. Kräutler, S. J. Lippard, R. Liskamp, K. Müller, E. M. Nolan, B. Samorì, G. Schneider, S. L. Schreiber, H. Schwalbe, C. Toniolo, C. A. A. van Boeckel, H. Waldmann and C. T. Walsh, ChemBioChem, 2009, 10, 16–29 CrossRef CAS.
- S. L. Schreiber, Nat. Chem. Biol., 2005, 1, 64–66 CrossRef CAS.
- Y. Li, C. R. Geyer and D. Sen, Biochemistry, 1996, 35, 6911–6922 CrossRef CAS.
- Q. Chen, I. D. Kuntz and R. H. Shafer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2635–2639 CrossRef CAS.
- D. Sun, B. Thompson, B. E. Cathers, M. Salazar, S. M. Kerwin, J. O. Trent, T. C. Jenkins, S. Neidle and L. H. Hurley, J. Med. Chem., 1997, 40, 2113–2116 CrossRef CAS.
- S. Müller and R. Rodriguez, Expert Rev. Clin. Pharmacol., 2014, 7, 663–679 CrossRef.
- F. Raguseo, S. Chowdhury, A. Minard and M. Di Antonio, Chem. Commun., 2020, 56, 1317–1324 RSC.
- D. Monchaud, Annu. Rep. Med. Chem., 2020, 54, 133–160 Search PubMed.
- D. Monchaud and M. P. Teulade-Fichou, Org. Biomol. Chem., 2008, 6, 627–636 RSC.
- S. A. Ohnmacht and S. Neidle, Bioorg. Med. Chem. Lett., 2014, 24, 2602–2612 CrossRef CAS.
- W. J. Chung, B. Heddi, M. Tera, K. Iida, K. Nagasawa and A. T. Phan, J. Am. Chem. Soc., 2013, 135, 13495–13501 CrossRef CAS.
- W. J. Chung, B. Heddi, F. Hamon, M. P. Teulade-Fichou and A. T. Phan, Angew. Chem., Int. Ed., 2014, 53, 999–1002 CrossRef CAS.
- A. T. Phan, V. Kuryavyi, H. Y. Gaw and D. J. Patel, Nat. Chem. Biol., 2005, 1, 167–173 CrossRef CAS.
- P. Murat, Y. Singh and E. Defrancq, Chem. Soc. Rev., 2011, 40, 5293–5307 RSC.
- J. Jaumot and R. Gargallo, Curr. Pharm. Des., 2012, 18, 1900–1916 CrossRef CAS.
- K. M. Felsenstein, L. B. Saunders, J. K. Simmons, E. Leon, D. R. Calabrese, S. Zhang, A. Michalowski, P. Gareiss, B. A. Mock and J. S. Schneekloth, ACS Chem. Biol., 2016, 11, 138–148 CrossRef.
- S. Ray, D. Tillo, R. E. Boer, N. Assad, M. Barshai, G. Wu, Y. Orenstein, D. Yang, J. S. Schneekloth, Jr. and C. Vinson, ACS Chem. Biol., 2020, 15, 925–935 CrossRef CAS.
- B. L. Allen-Petersen and R. C. Sears, BioDrugs, 2019, 33, 539–553 CrossRef.
- K. Shin-ya, K. Wierzba, K. Matsuo, T. Ohtani, Y. Yamada, K. Furihata, Y. Hayakawa and H. Seto, J. Am. Chem. Soc., 2001, 123, 1262–1263 CrossRef CAS.
- T. Tauchi, K. Shin-Ya, G. Sashida, M. Sumi, A. Nakajima, T. Shimamoto, J. H. Ohyashiki and K. Ohyashiki, Oncogene, 2003, 22, 5338–5347 CrossRef CAS.
- N. Temime-Smaali, L. Guittat, A. Sidibe, K. Shin-Ya, C. Trentesaux and J. F. Riou, PLoS One, 2009, 4, e6919 CrossRef.
- T. Miyazaki, Y. Pan, K. Joshi, D. Purohit, B. Hu, H. Demir, S. Mazumder, S. Okabe, T. Yamori, M. Viapiano, K. Shin-ya, H. Seimiya and I. Nakano, Clin. Cancer Res., 2012, 18, 1268–1280 CrossRef CAS.
- D. Gomez, T. Wenner, B. Brassart, C. Douarre, M. F. O’Donohue, V. El Khoury, K. Shin-Ya, H. Morjani, C. Trentesaux and J. F. Riou, J. Biol. Chem., 2006, 281, 38721–38729 CrossRef CAS.
- S. L. Palumbo, R. M. Memmott, D. J. Uribe, Y. Krotova-Khan, L. H. Hurley and S. W. Ebbinghaus, Nucleic Acids Res., 2008, 36, 1755–1769 CrossRef CAS.
- M. Tera, H. Ishizuka, M. Takagi, M. Suganuma, K. Shin-Ya and K. Nagasawa, Angew. Chem., Int. Ed., 2008, 47, 5557–5560 CrossRef CAS.
- T. Nakamura, S. Okabe, H. Yoshida, K. Iida, Y. Ma, S. Sasaki, T. Yamori, K. Shin-Ya, I. Nakano, K. Nagasawa and H. Seimiya, Sci. Rep., 2017, 7, 3605 CrossRef.
- E. Gavathiotis, R. A. Heald, M. F. G. Stevens and M. S. Searle, Angew. Chem., Int. Ed., 2001, 40, 4749 CrossRef CAS.
- C. Leonetti, S. Amodei, C. D. Angelo, A. Rizzo, B. Benassi, A. Antonelli, R. Elli, M. F. G. Stevens, M. D. Incalci, G. Zupi and A. Biroccio, Mol. Pharmacol., 2004, 66, 1138–1146 CrossRef CAS.
- E. Salvati, C. Leonetti, A. Rizzo, M. Scarsella, M. Mottolese, R. Galati, I. Sperduti, M. F. G. Stevens, M. D’Incalci, M. Blasco, G. Chiorino, S. Bauwens, B. Horard, E. Gilson, A. Stoppacciaro, G. Zupi and A. Biroccio, J. Clin. Invest., 2007, 117, 3236–3247 CrossRef CAS.
- A. Rizzo, E. Salvati, M. Porru, C. D’Angelo, M. F. Stevens, M. D’Incalci, C. Leonetti, E. Gilson, G. Zupi and A. Biroccio, Nucleic Acids Res., 2009, 37, 5353–5364 CrossRef CAS.
- A. De Cian, G. Cristofari, P. Reichenbach, E. De Lemos, D. Monchaud, M. P. Teulade-Fichou, K. Shin-ya, L. Lacroix, J. Lingner and J. L. Mergny, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17347–17352 CrossRef CAS.
- C. Granotier, G. Pennarun, L. Riou, F. Hoffschir, L. R. Gauthier, A. De Cian, D. Gomez, E. Mandine, J. F. Riou, J. L. Mergny, P. Mailliet, B. Dutrillaux and F. D. Boussin, Nucleic Acids Res., 2005, 33, 4182–4190 CrossRef CAS.
- G. Pennarun, C. Granotier, F. Hoffschir, E. Mandine, D. Biard, L. R. Gauthier and F. D. Boussin, Nucleic Acids Res., 2008, 36, 1741–1754 CrossRef CAS.
- G. Pennarun, F. Hoffschir, D. Revaud, C. Granotier, L. R. Gauthier, P. Mailliet, D. S. Biard and F. D. Boussin, Nucleic Acids Res., 2010, 38, 2955–2963 CrossRef CAS.
- L. R. Gauthier, C. Granotier, F. Hoffschir, O. Etienne, A. Ayouaz, C. Desmaze, P. Mailliet, D. S. Biard and F. D. Boussin, Cell. Mol. Life Sci., 2012, 69, 629–640 CrossRef CAS.
- A. De Cian, E. DeLemos, J. L. Mergny, M. P. Teulade-Fichou and D. Monchaud, J. Am. Chem. Soc., 2007, 129, 1856–1857 CrossRef CAS.
- J. Lopes, A. Piazza, R. Bermejo, B. Kriegsman, A. Colosio, M. P. Teulade-Fichou, M. Foiani and A. Nicolas, EMBO J., 2011, 30, 4033–4046 CrossRef CAS.
- J. Zimmer, E. M. C. Tacconi, C. Folio, S. Badie, M. Porru, K. Klare, M. Tumiati, E. Markkanen, S. Halder, A. Ryan, S. P. Jackson, K. Ramadan, S. G. Kuznetsov, A. Biroccio, J. E. Sale and M. Tarsounas, Mol. Cell, 2016, 61, 449–460 CrossRef CAS.
- K. Zyner, D. S. Mulhearn, S. Adhikari, S. M. Cuesta, M. Di Antonio, N. Erard, G. J. Hannon, D. Tannahill and S. Balasubramanian, eLife, 2019, 8, e46793 CrossRef.
- R. Rodriguez, S. Müller, J. A. Yeoman, C. Trentesaux, J. F. Riou and S. Balasubramanian, J. Am. Chem. Soc., 2008, 130, 15758–15759 CrossRef CAS.
- D. Koirala, S. Dhakal, B. Ashbridge, Y. Sannohe, R. Rodriguez, H. Sugiyama, S. Balasubramanian and H. Mao, Nat. Chem., 2011, 3, 782–787 CrossRef CAS.
- M. Di Antonio, K. I. E. McLuckie and S. Balasubramanian, J. Am. Chem. Soc., 2014, 136, 5860–5863 CrossRef CAS.
- S. Müller, S. Kumari, R. Rodriguez and S. Balasubramanian, Nat. Chem., 2010, 2, 1095–1098 CrossRef.
- T. Cañeque, S. Müller and R. Rodriguez, Nat. Rev. Chem., 2018, 2, 202–215 CrossRef.
- L. C. Kim, L. Song and E. B. Haura, Nat. Rev. Clin. Oncol., 2009, 6, 587–595 CrossRef CAS.
- J. F. Moruno-Manchon, E. C. Koellhoffer, J. Gopakumar, S. Hambarde, N. Kim, L. D. McCullough and A. S. Tsvetkov, Aging, 2017, 9, 1957–1970 CrossRef CAS.
- W. Duan, A. Rangan, H. Vankayalapati, M. Y. Kim, Q. Zeng, D. Sun, H. Han, O. Y. Fedoroff, D. Nishioka, S. Y. Rha, E. Izbicka, D. D. Von Hoff and L. H. Hurley, Mol. Cancer Ther., 2001, 1, 103–120 CAS.
- D. Drygin, A. Siddiqui-Jain, S. O’Brien, M. Schwaebe, A. Lin, J. Bliesath, C. B. Ho, C. Proffitt, K. Trent, J. P. Whitten, J. K. C. Lim, D. Von Hoff, K. Anderes and W. G. Rice, Cancer Res., 2009, 69, 7653–7661 CrossRef CAS.
- M. Haddach, M. K. Schwaebe, J. Michaux, J. Nagasawa, S. E. O’Brien, J. P. Whitten, F. Pierre, P. Kerdoncuff, L. Darjania, R. Stansfield, D. Drygin, K. Anderes, C. Proffitt, J. Bliesath, A. Siddiqui-Jain, M. Omori, N. Huser, W. G. Rice and D. M. Ryckman, ACS Med. Chem. Lett., 2012, 3, 602–606 CrossRef CAS.
- D. Drygin, A. Lin, J. Bliesath, C. B. Ho, S. E. O’Brien, C. Proffitt, M. Omori, M. Haddach, M. K. Schwaebe, A. Siddiqui-Jain, N. Streiner, J. E. Quin, E. Sanij, M. J. Bywater, R. D. Hannan, D. Ryckman, K. Anderes and W. G. Rice, Cancer Res., 2011, 71, 1418–1430 CrossRef CAS.
- S. S. Negi and P. Brown, Oncotarget, 2015, 6, 18094–18104 CrossRef.
- E. Sanij, K. M. Hannan, J. Xuan, S. Yan, J. E. Ahern, A. S. Trigos, N. Brajanovski, J. Son, K. T. Chan, O. Kondrashova, E. Lieschke, M. J. Wakefield, D. Frank, S. Ellis, C. Cullinane, J. Kang, G. Poortinga, P. Nag, A. J. Deans, K. K. Khanna, L. Mileshkin, G. A. McArthur, J. Soong, E. M. J. J. Berns, R. D. Hannan, C. L. Scott, K. E. Sheppard and R. B. Pearson, Nat. Commun., 2020, 11, 2641 CrossRef CAS.
- H. Xu, M. Di Antonio, S. McKinney, V. Mathew, B. Ho, N. J. O’Neil, N. Dos Santos, J. Silvester, V. Wei, J. Garcia, F. Kabeer, D. Lai, P. Soriano, J. Banáth, D. S. Chiu, D. Yap, D. D. Le, F. B. Ye, A. Zhang, K. Thu, J. Soong, S. C. Lin, A. H. C. Tsai, T. Osako, T. Algara, D. N. Saunders, J. Wong, J. Xian, M. B. Bally, J. D. Brenton, G. W. Brown, S. P. Shah, D. Cescon, T. W. Mak, C. Caldas, P. C. Stirling, P. Hieter, S. Balasubramanian and S. Aparicio, Nat. Commun., 2017, 8, 14432 CrossRef CAS.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS.
- P. M. Bruno, M. Lu, K. A. Dennis, H. Inam, C. J. Moore, J. Sheehe, S. J. Elledge, M. T. Hemann and J. R. Pritchard, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4053–4060 CrossRef CAS.
- J. Beauvarlet, P. Bensadoun, E. Darbo, G. Labrunie, B. Rousseau, E. Richard, I. Draskovic, A. Londono-Vallejo, J. W. Dupuy, R. N. Das, A. Guédin, G. Robert, F. Orange, S. Croce, V. Valesco, P. Soubeyran, K. M. Ryan, J. L. Mergny and M. Djavaheri-Mergny, Nucleic Acids Res., 2019, 47, 2739–2756 CrossRef CAS.
- W. J. Zhou, R. Deng, X. Y. Zhang, G. K. Feng, L. Q. Gu and X. F. Zhu, Mol. Cancer Ther., 2009, 8, 3203–3213 CrossRef CAS.
- A. De Magis, S. G. Manzo, M. Russo, J. Marinello, R. Morigi, O. Sordet and G. Capranico, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 816–825 CrossRef CAS.
- A. Thomas and Y. Pommier, Clin. Cancer Res., 2019, 25, 6581–6589 CrossRef CAS.
- B. L. Staker, M. D. Feese, M. Cushman, Y. Pommier, D. Zembower, L. Stewart and A. B. Burgin, J. Med. Chem., 2005, 48, 2336–2345 CrossRef CAS.
- A. Henderson, Y. Wu, Y. C. Huang, E. A. Chavez, J. Platt, F. B. Johnson, R. M. Brosh, D. Sen and P. Lansdorp, Nucleic Acids Res., 2014, 42, 860–869 CrossRef CAS.
- D. C. Odermatt, W. Ting, C. L. Id, S. Wild, S. K. J. Id, E. Rothenberg and K. G. Id, PLoS Genet., 2020, 16, e1008740 CrossRef CAS.
- L. A. Watson, L. A. Solomon, J. R. Li, Y. Jiang, M. Edwards, K. Shin-Ya, F. Beier and N. G. Bérubé, J. Clin. Invest., 2013, 123, 2049–2063 CrossRef CAS.
- K. I. E. McLuckie, M. Di Antonio, H. Zecchini, J. Xian, C. Caldas, B. Krippendor, D. Tannahill, C. Lowe and S. Balasubramanian, J. Am. Chem. Soc., 2013, 135, 9640–9643 CrossRef CAS.
- S. Li, H. Lu, Z. Wang, Q. Hu, H. Wang, R. Xiang, T. Chiba and X. Wu, iScience, 2019, 16, 63–78 CrossRef CAS.
- W. Feng, D. A. Simpson, J. Carvajal-Garcia, B. A. Price, R. J. Kumar, L. E. Mose, R. D. Wood, N. Rashid, J. E. Purvis, J. S. Parker, D. A. Ramsden and G. P. Gupta, Nat. Commun., 2019, 10, 4286 CrossRef.
- M. Aggarwal, J. A. Sommers, R. H. Shoemaker and R. M. Brosh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1525–1530 CrossRef CAS.
- P. Phatak, J. C. Cookson, F. Dai, V. Smith, R. B. Gartenhaus, M. F. G. Stevens and A. M. Burger, Br. J. Cancer, 2007, 96, 1223–1233 CrossRef CAS.
- C. Leonetti, M. Scarsella, G. Riggio, A. Rizzo, E. Salvati, M. D’Incalci, L. Staszewsky, R. Frapolli, M. F. Stevens, A. Stoppacciaro, M. Mottolese, B. Antoniani, E. Gilson, G. Zupi and A. Biroccio, Clin. Cancer Res., 2008, 14, 7284–7291 CrossRef CAS.
- E. Salvati, M. Scarsella, M. Porru, A. Rizzo, S. Iachettini, L. Tentori, G. Graziani, M. D’Incalci, M. F. G. Stevens, A. Orlandi, D. Passeri, E. Gilson, G. Zupi, C. Leonetti and A. Biroccio, Oncogene, 2010, 29, 6280–6293 CrossRef CAS.
- A. Biroccio, M. Porru, A. Rizzo, E. Salvati, C. D’Angelo, A. Orlandi, D. Passeri, M. Franceschin, M. F. G. Stevens, E. Gilson, G. Beretta, G. Zupi, C. Pisano, F. Zunino and C. Leonetti, Clin. Cancer Res., 2011, 17, 2227–2236 CrossRef CAS.
- A. Rizzo, S. Iachettini, P. Zizza, C. Cingolani, M. Porru, S. Artuso, M. Stevens, M. Hummersone, A. Biroccio, E. Salvati and C. Leonetti, J. Exp. Clin. Cancer Res., 2015, 33, 81 CrossRef.
- F. Berardinelli, M. Tanori, D. Muoio, M. Buccarelli, A. Di Masi, S. Leone, L. Ricci-Vitiani, R. Pallini, M. Mancuso and A. Antoccia, J. Exp. Clin. Cancer Res., 2019, 38, 311 CrossRef CAS.
- F. Berardinelli, S. Siteni, C. Tanzarella, M. F. Stevens, A. Sgura and A. Antoccia, DNA Repair, 2015, 25, 104–115 CrossRef CAS.
- Y. Wang, J. Yang, A. T. Wild, W. H. Wu, R. Shah, C. Danussi, G. J. Riggins, K. Kannan, E. P. Sulman, T. A. Chan and J. T. Huse, Nat. Commun., 2019, 10, 943 CrossRef.
- M. Porru, P. Zizza, M. Franceschin, C. Leonetti and A. Biroccio, Biochim. Biophys. Acta, 2017, 1861, 1362–1370 CrossRef CAS.
- T. C. Chou and P. Talalay, Adv. Enzyme Regul., 1984, 22, 27–55 CrossRef CAS.
- T. C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS.
- C. I. Bliss, Ann. Appl. Biol., 1939, 26, 585–615 CrossRef CAS.
- R. Rodriguez and K. M. Miller, Nat. Rev. Genet., 2014, 15, 783–796 CrossRef CAS.
- S. F. Cai, C. W. Chen and S. A. Armstrong, Mol. Cell, 2015, 60, 561–570 CrossRef CAS.
- Y. Cheng, C. He, M. Wang, X. Ma, F. Mo, S. Yang, J. Han and X. Wei, Signal Transduction Targeted Ther., 2019, 4, 62 CrossRef.
- D. S. Dizon, L. Damstrup, N. J. Finkler, U. Lassen, P. Celano, R. Glasspool, E. Crowley, H. S. Lichenstein, P. Knoblach and R. T. Penson, Int. J. Gynecol. Cancer, 2012, 22, 979–986 CrossRef.
- G. Wang and K. M. Vasquez, DNA Repair, 2014, 19, 143–151 CrossRef CAS.
- T. Kato, H. Kurahashi and B. S. Emanuel, Curr. Opin. Genet. Dev., 2012, 22, 221–228 CrossRef CAS.
- T. J. Treangen and S. L. Salzberg, Nat. Rev. Genet., 2012, 13, 36–46 CrossRef CAS.
- J. Choi and T. Majima, Chem. Soc. Rev., 2011, 40, 5893–5909 RSC.
- S. Kaushal and C. H. Freudenreich, Genes, Chromosomes Cancer, 2018, 58, 270–283 Search PubMed.
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 CrossRef CAS.
- S. Ravichandran, V. K. Subramani and K. K. Kim, Biophys. Rev., 2019, 11, 383–387 CrossRef CAS.
- A. Aguilera and B. Gómez-González, Nat. Struct. Mol. Biol., 2017, 24, 439–443 CrossRef CAS.
- F. Chedin, C. J. Benham and K. Musier-Forsyth, J. Biol. Chem., 2020, 295, 4684–4695 CrossRef CAS.
- B. Gómez-González and A. Aguilera, Genes Dev., 2019, 33, 1008–1026 CrossRef.
- C. Niehrs and B. Luke, Nat. Rev. Mol. Cell Biol., 2020, 21, 167–178 CrossRef CAS.
- M. Duca, P. Vekhoff, K. Oussedik, L. Halby and P. B. Arimondo, Nucleic Acids Res., 2008, 36, 5123–5138 CrossRef CAS.
- M. Thomas, R. L. White and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 2294–2298 CrossRef CAS.
- M. P. Crossley, M. Bocek and K. A. Cimprich, Mol. Cell, 2019, 73, 398–411 CrossRef CAS.
- L. A. Sanz and F. Chédin, Nat. Protoc., 2019, 14, 1734–1755 CrossRef CAS.
- P. A. Ginno, P. L. Lott, H. C. Christensen, I. Korf and F. Chédin, Mol. Cell, 2012, 45, 814–825 CrossRef CAS.
- K. J. Abraham, N. Khosraviani, J. N. Y. Chan, A. Gorthi, A. Samman, D. Y. Zhao, M. Wang, M. Bokros, E. Vidya, L. A. Ostrowski, R. Oshidari, V. Pietrobon, P. S. Patel, A. Algouneh, R. Singhania, Y. Liu, V. T. Yerlici, D. D. De Carvalho, M. Ohh, B. C. Dickson, R. Hakem, J. F. Greenblatt, S. Lee, A. J. R. Bishop and K. Mekhail, Nature, 2020, 585, 298–302 CrossRef CAS.
- M. R. Lieber, Annu. Rev. Biochem., 2010, 79, 181–211 CrossRef CAS.
- S. Britton, E. Dernoncourt, C. Delteil, C. Froment, O. Schiltz, B. Salles, P. Frit and P. Calsou, Nucleic Acids Res., 2014, 42, 9047–9062 CrossRef CAS.
- S. Tuduri, L. Crabbé, C. Conti, H. Tourrière, H. Holtgreve-Grez, A. Jauch, V. Pantesco, J. De Vos, A. Thomas, C. Theillet, Y. Pommier, J. Tazi, A. Coquelle and P. Pasero, Nat. Cell Biol., 2009, 11, 1315–1324 CrossRef CAS.
- J. Tazi, N. Bakkour, J. Soret, L. Zekri, B. Hazra, W. Laine, B. Baldeyrou, A. Lansiaux and C. Bailly, Mol. Pharmacol., 2005, 67, 1186–1194 CrossRef CAS.
- A. De Magis, S. G. Manzo, M. Russo, J. Marinello, R. Morigi, O. Sordet and G. Capranico, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 816–825 CrossRef CAS.
- J. Amato, G. Miglietta, R. Morigi, N. Iaccarino, A. Locatelli, A. Leoni, E. Novellino, B. Pagano, G. Capranico and A. Randazzo, J. Med. Chem., 2020, 63, 3090–3103 CrossRef CAS.
- D. M. J. Lilley, Q. Rev. Biophys., 2000, 33, 109–159 CrossRef CAS.
- V. Brázda, J. Kolomazník, J. Lýsek, L. Hároníková, J. Coufal and J. Št’astný, Biochem. Biophys. Res. Commun., 2016, 478, 1739–1745 CrossRef.
- E. M. Strawbridge, G. Benson, Y. Gelfand and C. J. Benham, Curr. Genet., 2010, 56, 321–340 CrossRef CAS.
- M. Čutová, J. Manta, O. Porubiaková, P. Kaura, J. Šťastný, E. B. Jagelská, P. Goswami, M. Bartas and V. Brázda, Genomics, 2020, 112, 1897–1901 CrossRef.
- A. Bedrat, L. Lacroix and J. L. Mergny, Nucleic Acids Res., 2016, 44, 1746–1759 CrossRef.
- R. Cox and S. M. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 5237–5242 CrossRef CAS.
- X. Lu, G. Wang, A. Bacolla, J. Zhao, S. Spitser and K. M. Vasquez, Cell Rep., 2015, 10, 1674–1680 CrossRef.
- Y. Wang and F. C. C. Leung, FEBS J., 2009, 276, 1986–1998 CrossRef CAS.
- D. G. Albertson, Trends Genet., 2006, 22, 447–455 CrossRef CAS.
- A. Aguilera and T. García-Muse, Annu. Rev. Genet., 2013, 47, 1–32 CrossRef CAS.
- A. B. Reams and J. R. Roth, Cold Spring Harbor Perspect. Biol., 2015, 7, a016592 CrossRef.
- C. T. McMurray, Nat. Rev. Genet., 2010, 11, 786–799 CrossRef CAS.
- W. K. Chu and I. D. Hickson, Nat. Rev. Cancer, 2009, 9, 644–654 CrossRef CAS.
- I. Voineagu, V. Narayanan, K. S. Lobachev and S. M. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9936–9941 CrossRef CAS.
- I. Voineagu, C. F. Surka, A. A. Shishkin, M. M. Krasilnikova and S. M. Mirkin, Nat. Struct. Mol. Biol., 2009, 16, 226–228 CrossRef CAS.
- N. Shastri, Y. C. Tsai, S. Hile, D. Jordan, B. Powell, J. Chen, D. Maloney, M. Dose, Y. Lo, T. Anastassiadis, O. Rivera, T. Kim, S. Shah, P. Borole, K. Asija, X. Wang, K. D. Smith, D. Finn, J. Schug, R. Casellas, L. A. Yatsunyk, K. A. Eckert and E. J. Brown, Mol. Cell, 2018, 72, 222–238 CrossRef CAS.
- L. Gellon, S. Kaushal, J. Cebrián, M. Lahiri, S. M. Mirkin and C. H. Freudenreich, Nucleic Acids Res., 2019, 47, 794–805 CrossRef CAS.
- M. L. Nicolette, K. Lee, Z. Guo, M. Rani, J. M. Chow, S. E. Lee and T. T. Paull, Nat. Struct. Mol. Biol., 2010, 17, 1478–1485 CrossRef CAS.
- K. S. Lobachev, D. A. Gordenin and M. A. Resnick, Cell, 2002, 108, 183–193 CrossRef CAS.
- M. McVey and S. E. Lee, Trends Genet., 2008, 24, 529–538 CrossRef CAS.
- A. L. Brogden, N. H. Hopcroft, M. Searcey and C. J. Cardin, Angew. Chem., Int. Ed., 2007, 46, 3850–3854 CrossRef CAS.
- A. Oleksi, A. G. Blanco, R. Boer, I. Usón, J. Aymamí, A. Rodger, M. J. Hannon and M. Coll, Angew. Chem., Int. Ed., 2006, 45, 1227–1231 CrossRef CAS.
- Q. Guo, N. C. Seeman and N. R. Kallenbach, Biochemistry, 1989, 28, 2355–2359 CrossRef CAS.
- M. Lu, Q. Guo and N. R. Kallenbach, Crit. Rev. Biochem. Mol. Biol., 1992, 27, 157–190 CrossRef CAS.
- M. Lu, Q. Guo, R. F. Pasternack, D. J. Wink, N. C. Seeman and N. R. Kallenbach, Biochemistry, 1990, 29, 1614–1624 CrossRef.
- M. N. Stojanovic, P. De Prada and D. W. Landry, J. Am. Chem. Soc., 2000, 122, 11547–11548 CrossRef CAS.
- M. N. Stojanovic, P. de Prada and D. W. Landry, J. Am. Chem. Soc., 2001, 123, 4928–4931 CrossRef CAS.
- M. N. Stojanovic, E. G. Green, S. Semova, D. B. Nikić and D. W. Landry, J. Am. Chem. Soc., 2003, 125, 6085–6089 CrossRef CAS.
- M. N. Stojanovic and T. S. Worgall, Curr. Opin. Chem. Biol., 2010, 14, 751–757 CrossRef CAS.
- M. Lu, Q. Guo, N. C. Seeman and N. R. Kallenbach, Biochemistry, 1990, 29, 3407–3412 CrossRef CAS.
- K. V. Kepple, J. L. Boldt and A. M. Segall, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6867–6872 CrossRef CAS.
- K. V. Kepple, N. Patel, P. Salamon and A. M. Segall, Nucleic Acids Res., 2008, 36, 5319–5334 CrossRef CAS.
- M. L. Bolla, E. V. Azevedo, J. M. Smith, R. E. Taylor, D. K. Ranjit, A. M. Segall and S. R. McAlpine, Org. Lett., 2003, 5, 109–112 CrossRef CAS.
- P. S. Pan, F. A. Curtis, C. L. Carroll, I. Medina, L. A. Liotta, G. J. Sharples and S. R. McAlpine, Bioorg. Med. Chem., 2006, 14, 4731–4739 CrossRef CAS.
- L. A. Howell, R. A. Bowater, M. A. O’Connell, A. P. Reszka, S. Neidle and M. Searcey, ChemMedChem, 2012, 7, 792–804 CrossRef CAS.
- L. A. Howell, Z. A. E. Waller, R. Bowater, M. A. O’Connell and M. Searcey, Chem. Commun., 2011, 47, 8262–8264 RSC.
- D. R. Boer, J. M. C. A. Kerckhoffs, Y. Parajo, M. Pascu, I. Usón, P. Lincoln, M. J. Hannon and M. Coll, Angew. Chem., Int. Ed., 2010, 49, 2336–2339 CrossRef CAS.
- I. Gamba, G. Rama, E. Ortega-Carrasco, J. D. Maréchal, J. Martínez-Costas, M. Eugenio Vázquez and M. V. López, Chem. Commun., 2014, 50, 11097–11100 RSC.
- S. Vuong, L. Stefan, P. Lejault, Y. Rousselin, F. Denat and D. Monchaud, Biochimie, 2012, 94, 442–450 CrossRef CAS.
- L. Stefan, B. Bertrand, P. Richard, P. Le Gendre, F. Denat, M. Picquet and D. Monchaud, ChemBioChem, 2012, 13, 1905–1912 CrossRef CAS.
- S. A. Barros and D. M. Chenoweth, Angew. Chem., Int. Ed., 2014, 53, 13746–13750 CrossRef CAS.
- S. A. Barros and D. M. Chenoweth, Chem. Sci., 2015, 6, 4752–4755 RSC.
- J. Novotna, A. Laguerre, A. Granzhan, M. Pirrotta, M. P. Teulade-Fichou and D. Monchaud, Org. Biomol. Chem., 2015, 13, 215–222 RSC.
- L. Guyon, M. Pirrotta, K. Duskova, A. Granzhan, M. P. Teulade-Fichou and D. Monchaud, Nucleic Acids Res., 2018, 46, e16 CrossRef.
- K. Duskova, J. Lamarche, S. Amor, C. Caron, N. Queyriaux, M. Gaschard, M. J. Penouilh, G. De Robillard, D. Delmas, C. H. Devillers, A. Granzhan, M. P. Teulade-Fichou, M. Chavarot-Kerlidou, B. Therrien, S. Britton and D. Monchaud, J. Med. Chem., 2019, 62, 4456–4466 CrossRef CAS.
- J. Zhu, C. J. E. Haynes, M. Kieffer, J. L. Greenfield, R. D. Greenhalgh, J. R. Nitschke and U. F. Keyser, J. Am. Chem. Soc., 2019, 141, 11358–11362 CrossRef CAS.
- Z. Yang, Y. Chen, G. Li, Z. Tian, L. Zhao, X. Wu, Q. Ma, M. Liu and P. Yang, Chem. – Eur. J., 2018, 24, 6087–6093 CrossRef CAS.
- J. Xia, Q. Mei and S. M. Rosenberg, Trends Genet., 2019, 35, 383–395 CrossRef CAS.
- J. Xia, L. T. Chen, Q. Mei, C. H. Ma, J. A. Halliday, H. Y. Lin, D. Magnan, J. P. Pribis, D. M. Fitzgerald, H. M. Hamilton, M. Richters, R. B. Nehring, X. Shen, L. Li, D. Bates, P. J. Hastings, C. Herman, M. Jayaram and S. M. Rosenberg, Sci. Adv., 2016, 2, e1601605 CrossRef.
- K. Ghosh, K. L. Chi, F. Guo, A. M. Segall and G. D. Van Duyne, J. Biol. Chem., 2005, 280, 8290–8299 CrossRef CAS.
- C. W. Gunderson and A. M. Segall, Mol. Microbiol., 2006, 59, 1129–1148 CrossRef CAS.
- M. Dey, S. Patra, L. Y. Su and A. M. Segall, PLoS One, 2013, 8, e78751 CrossRef.
- A. C. G. Hotze, N. J. Hodges, R. E. Hayden, C. Sanchez-Cano, C. Paines, N. Male, M. K. Tse, C. M. Bunce, J. K. Chipman and M. J. Hannon, Chem. Biol., 2008, 15, 1258–1267 CrossRef CAS.
- C. Ducani, A. Leczkowska, N. J. Hodges and M. J. Hannon, Angew. Chem., Int. Ed., 2010, 49, 8942–8945 CrossRef CAS.
- G. I. Pascu, A. C. G. Hotze, C. Sanchez-Cano, B. M. Kariuki and M. J. Hannon, Angew. Chem., Int. Ed., 2007, 46, 4374–4378 CrossRef CAS.
- U. McDonnell, J. M. C. A. Kerchoffs, R. P. M. Castineiras, M. R. Hicks, A. C. G. Hotze, M. J. Hannon and A. Rodger, Dalton Trans., 2008, 667–675 RSC.
- H. Yu, X. Wang, M. Fu, J. Ren and X. Qu, Nucleic Acids Res., 2008, 36, 5695–5703 CrossRef CAS.
- C. Zhao, J. Geng, L. Feng, J. Ren and X. Qu, Chem. – Eur. J., 2011, 17, 8209–8215 CrossRef CAS.
- C. Zhao, L. Wu, J. Ren, Y. Xu and X. Qu, J. Am. Chem. Soc., 2013, 135, 18786–18789 CrossRef CAS.
- V. Brabec, S. E. Howson, R. A. Kaner, R. M. Lord, J. Malina, R. M. Phillips, Q. M. A. Abdallah, P. C. McGowan, A. Rodger and P. Scott, Chem. Sci., 2013, 4, 4407–4416 RSC.
- A. Granzhan, N. Kotera and M. P. Teulade-Fichou, Chem. Soc. Rev., 2014, 43, 3630–3665 RSC.
- N. Kotera, A. Granzhan and M. P. Teulade-Fichou, Biochimie, 2016, 128–129, 133–137 CrossRef CAS.
- N. Kotera, F. Poyer, A. Granzhan and M. P. Teulade-Fichou, Chem. Commun., 2015, 51, 15948–15951 RSC.
- C. Caron, X. N. T. Duong, R. Guillot, S. Bombard and A. Granzhan, Chem. – Eur. J., 2019, 25, 1949–1962 CrossRef CAS.
- K. Duskova, P. Lejault, É. Benchimol, R. Guillot, S. Britton, A. Granzhan and D. Monchaud, J. Am. Chem. Soc., 2020, 142, 424–435 CrossRef CAS.
- F. H. Crick, Symp. Soc. Exp. Biol., 1958, 12, 138–163 CAS.
- F. Crick, Nature, 1970, 227, 561–563 CrossRef CAS.
- F. Crick, Nature, 1974, 248, 766–769 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |