
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntibacterial activity of a dual peptide targeting the Escherichia coli sliding clamp and the ribosome†
Christophe
Andr釧
 a,
Florian
Veillard‡¶
b,
Philippe
Wolff
c,
Anne-Marie
Lobstein
c,
Guillaume
Compain
a,
Clément
Monsarrat
a,
Jean-Marc
Reichhart
b,
Camille
Noûs
d,
Dominique Y.
Burnouf
*c,
Gilles
Guichard
*a and
Jérôme E.
Wagner‡
*e
a,
Florian
Veillard‡¶
b,
Philippe
Wolff
c,
Anne-Marie
Lobstein
c,
Guillaume
Compain
a,
Clément
Monsarrat
a,
Jean-Marc
Reichhart
b,
Camille
Noûs
d,
Dominique Y.
Burnouf
*c,
Gilles
Guichard
*a and
Jérôme E.
Wagner‡
*e
aUniv. Bordeaux, CNRS, Bordeaux INP, CBMN, UMR 5248, Institut Européen de Chimie et Biologie, 2 rue Robert Escarpit, F-33607 Pessac, France. E-mail: g.guichard@iecb.u-bordeaux.fr
bInsect Models of Innate Immunity, UPR 9022-CNRS, Institut de Biologie Moléculaire et Cellulaire, 67000 Strasbourg, France
cCNRS, Architecture et Réactivité de l’ARN, UPR 9002, Institut de Biologie Moléculaire et Cellulaire, 67000 Strasbourg, France. E-mail: d.burnouf@ibmc-cnrs.unistra.fr
dLaboratoire Cogitamus, 1 3/4 rue Descartes, 75005 Paris, France
eUniversité de Strasbourg, CNRS, Biotechnologie et Signalisation Cellulaire, UMR 7242, Ecole Supérieure de Biotechnologie de Strasbourg, 67400 Illkirch, France. E-mail: jerome.wagner@unistra.fr
First published on 16th July 2020
Abstract
The bacterial processivity factor, or sliding clamp (SC), is a target of choice for new antibacterial drugs development. We have previously developed peptides that target Escherichia coli SC and block its interaction with DNA polymerases in vitro. Here, one such SC binding peptide was fused to a Proline-rich AntiMicrobial Peptide (PrAMP) to allow its internalization into E. coli cells. Co-immunoprecipitation assays with a N-terminally modified bifunctional peptide that still enters the bacteria but fails to interact with the bacterial ribosome, the major target of PrAMPs, demonstrate that it actually interacts with the bacterial SC. Moreover, when compared to SC non-binding controls, this peptide induces a ten-fold higher antibacterial activity against E. coli, showing that the observed antimicrobial activity is linked to SC binding. Finally, an unmodified bifunctional compound significantly increases the survival of Drosophila melanogaster flies challenged by an E. coli infection. Our study demonstrates the potential of PrAMPs to transport antibiotics into the bacterial cytoplasm and validates the development of drugs targeting the bacterial processivity factor of Gram-negative bacteria as a promising new class of antibiotics.
Introduction
Antimicrobial resistance has become an acute problem for public health and economy and threatens the progress of modern medicine. Bacterial antibiotic resistance is spreading rapidly, and last resort antibiotics have become ineffective for several infections, as exemplified by the emergence of carbapenem and colistin resistant Enterobacteriaceae. The World Health Organization has recently published a priority list of organisms1 against which new compounds must be developed to control their impact on human health. Indeed, future success in tackling antimicrobial resistance relies notably on the development of new antibiotics directed towards new cellular targets. Ideally, these new drugs should also limit the frequency of resistance emergence. In this context, the bacterial replication processivity factor, or sliding clamp (SC) is a promising target because of its central function in the bacterial replisome dynamics.2–4 This protein (DnaN) forms a homodimeric molecular hub, which interacts with many partners involved in bacterial DNA metabolism. In particular, all DNA polymerases interact with SC5via a short peptide motif, for which a consensus sequence (QL[S/D]LF) has been defined,6 and the hydrophobic pocket where these peptides interact7,8 has been recognized as a potential molecular target for new drug development.4 It is anticipated that blocking the interaction between SC and the replicative polymerase will lead to cell death. More interestingly, as the specialized, SOS-regulated DNA polymerases (PolII, IV and V) are required for mutagenesis, inhibition of their activities should limit the possibility for bacteria to develop new resistance. Actually, these polymerases have been shown to directly participate in the establishment of ciprofloxacin resistance in E. coli9 and the necessity to interact with the SC to perform efficient mutagenesis has been demonstrated.5,10We have previously used a structure-based strategy to design small peptides that can interact effectively within the pocket of the Escherichia coli SC (EcSC), compete with DNA polymerases in vitro and totally abolish the SC dependent replication process.3,8 However, a strong limitation in the further development of these peptides into antimicrobial agents is their limited ability to cross the bacterial cell membrane. To overcome this problem, we sought to combine these peptides with a delivery vector in a single molecule to achieve a more efficient cellular uptake. Proline rich AntiMicrobial Peptides (PrAMPs), found in invertebrates and vertebrates11,12 as part of the innate immune response, are transported into a large panel of Gram-negative bacterial cells by the inner membrane transporters SbmA and MdtM,13,14 and appear to be well suited as carriers to transfer the otherwise non-penetrating SC targeting peptides into the bacterial cell. There are only few precedents in the literature for the use of PrAMPs to transport active molecules inside bacteria and eukaryotic cells.15–17 PrAMPS are also considered as a promising novel class of antibiotics12,18 and the main cellular target that accounts for their biological activity, namely the peptide exit channel (PEC) of the bacterial ribosome, has been recently identified.19–21
The aim of this study was to deliver a SC binding peptide into bacterial cells, to subsequently demonstrate its capacity to engage its molecular target in cellulo, and to assess its antimicrobial activity in vitro and its protective potency in an E. coli infection model. For this purpose, we first evaluated whether the expression of a SC binding peptide within E. coli cells was toxic for the bacteria. We then conjugated peptide P7, a reference SC binding peptide (Table 1) that we previously identified while studying the details of the ligand–target interaction,22,23 to PrAMP sequences, namely Onc11224 and unglycosylated pyrrhocoricin (Py),25 to form two bifunctional peptides Onc112-P7 and Pyrrhocoricin-P7 (Py-P7), respectively (Table 1). We found that such a dual peptide accumulates into E. coli cytoplasm and interact with the SC target. The antimicrobial activity of such a penetrating P7 SC binding peptide is demonstrated for the first time against E. coli by using a N-terminal modified peptide conjugate that is still internalized but does not interact with the ribosome anymore. Finally, we show that the dual targeting of the EcSC and Ec70S with a bifunctional peptide yields an increased protective effect in an E. coli infection assay of the fruit fly D. Melanogaster.
| Name | Sequencea | M w | Targets | K D (nM) ± sdb |
|---|---|---|---|---|
| a X = Cha: cyclohexylalanine. r: (D)-R amino acid. Scr: scramble. FITC: fluorescein isothiocyanate. Pyrrhocoricin (Py) was purchased from Novopro. For all peptides the purity, as determined by HPLC (λ = 214 nm), was ≥98%. b K D for each target were determined by ITC at 30 °C. Data are means ± sd of at least 2 independent experiments (see Table SI.4, ESI). ni: no specific interaction (see SI.2–4, ESI for full thermodynamic data and analysis). | ||||
| P 7 | Ac-QXDLF-OH | 716.82 | Ec SC | 194 ± 58 |
| Ec 70S ribosome | ni | |||
| Py | VDKGSYLPRPTPPRPIYNRN-NH2 | 2339.70 | Ec SC | ni |
| Ec 70S ribosome | 15 ± 10 | |||
| Py-P 7 | VDKGSYLPRPTPPRPIYNRNGPRQXDLF-OH | 3307.82 | Ec SC | 120 ± 20 |
| Ec 70S ribosome | 37 ± 0.3 | |||
| Py-P 7 Scr | VDKGSYLPRPTPPRPIYNRNGPRXFQLD-OH | 3307.82 | Ec SC | ni |
| Ec 70S ribosome | 76 ± 20 | |||
| FITC-Py | FITC-KVDKGSYLPRPTPPRPIYNRN-NH2 | 2826.17 | Ec SC | ni |
| Ec 70S ribosome | >104 | |||
| FITC-Py-P7 | FITC-KVDKGSYLPRPTPPRPIYNRNGPRQXDLF-OH | 3794.30 | Ec SC | 300 ± 30 |
| Ec 70S ribosome | >104 | |||
| FITC-Py-P7Scr | FITC-KVDKGSYLPRPTPPRPIYNRNGPRXFQLD-OH | 3794.30 | Ec SC | ni |
| Ec 70S ribosome | >104 | |||
| Onc112 | VDKPPYLPRPRPPRrIYNr-NH2 | 2389.85 | Ec SC | ni |
| Ec 70S ribosome | 84 ± 20 | |||
| Onc112-P 7 | VDKPPYLPRPRPPRrIYNrNGPRQXDLF-OH | 3472.08 | Ec SC | 200 ± 0.1 |
| Ec 70S ribosome | 70 ± 10 | |||
| Onc112-P 7 Scr | VDKPPYLPRPRPPRrIYNrNGPRXFQLD-OH | 3472.08 | Ec SC | ni |
| Ec 70S ribosome | 100 ± 0.9 | |||
Experimental
Overexpression of sliding clamp binding peptides
Phosphorylated and hybridized oligos pairs 8Fup/8Flow and 8Aup/8Alow (8Fup: 5′-CATGGGCCCGCGTCAGCTGGATCTGTTTTAATGA; 8Flow: 5′-GATCTCATTAAAAACAGATCCAGCTGACGCGGGCC; 8Aup: 5′-CATGGGCCCGCGTGCGCTGGATGCAGCGTAATGA; 8Alow: 5′-GATCTCATTACGCTGCATCCAGCTGACGCGGGCC) coding respectively for peptides MGPRQLDLF and MGPRALDAA were cloned in fusion with GST in the NcoI and BamHI restriction sites of pET-M30 plasmid.26 The integrity of the cloned sequences was verified by sequencing (Eurofins GATC Biotech, Germany). Resulting plasmids pETM30-8F and pETM30-8A as well as the empty vector pETM30 were transformed into chemically competent BL21(DE3) pLys cells and subsequently used as described in the legend to Fig. 1.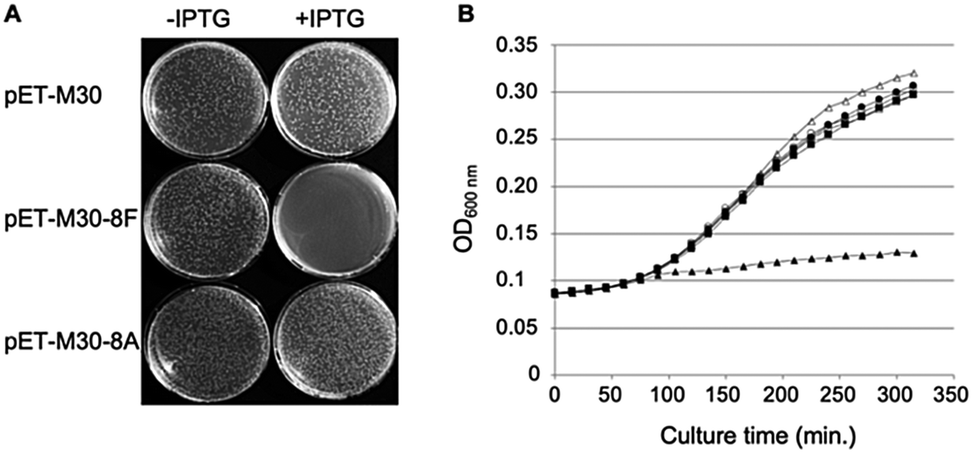 | ||
| Fig. 1 Expression of SC binding peptide in E. coli is toxic. (A) E. coli BL21 (DE3) pLysS cells were transformed with either pET-M30, pET-M30-8F or pET-M30-8A vectors expressing His-GST, and respectively the 8F SC binding peptide (MGPRQLDLF) or the SC non-binding mutant 8A (MGPRALDAA) in fusion with His-GST. After 1 hour incubation at 37 °C, one tenth of the transformation mixtures were plated on LB agar plates containing kanamycin, chloramphenicol and either 0 or 0.1 mM IPTG. The picture was taken after 18 hours incubation at 37 °C. (B) The growth of transformed E. coli BL21 (DE3) pLys cells in Luria broth complemented or not with IPTG was followed over a period of 5 h 30 min by measurement of OD600 every 15 minutes in a TECAN M200 plate reader. Open symbols: no IPTG in the medium; closed symbols: 0.5 mM IPTG; circles: empty vector (pETM30); squares: expression of SC non binding peptide (pETM30-8A); triangles: expression of SC binding peptide (pETM30-8F). | ||
Bacterial strains
E. coli strain D22 (F-, lpxC101, proA23, lac-28, tsx-81, trp-30, his-51, tufA1, rpsL173(strR), ampCp-1) was purchased at the E. coli Genetic Stock Center (Yale University) and used in the determination of MIC and in the peptide penetration assays. E. coli ATCC23724 cells were used to infect D. melanogaster flies. 70S ribosomes were purified from the E. coli MRE600 strain (Accession AY140951) and BL21 (DE3) pLys strain was used for production of the DnaN protein.Peptides synthesis
Commercially available reagents were used throughout without purification. N,N-Dimethylformamide (DMF, peptide synthesis-quality grade) was purchased from Carlo Erba, and piperidine and trifluoroacetic acid (TFA) were purchased from Alfa Aesar. Rink amide PS or Wang resin was purchased from MerckMillipore. O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU), N,N′-diisopropylcarbodiimide (DIC) and (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) and all standard N-Fmoc-protected L- and D-amino acids were purchased from Iris Biotech. N,N-Diisopropylethylamine (DIEA) and 5(6)-carboxyfluorescein (FITC) were purchased from Sigma-Aldrich. Peptides were synthesized on solid support from a fully automated microwave peptide synthesizer: Liberty Blue System synthesizer (CEM Corporation). RP-HPLC-quality acetonitrile (CH3CN, Sigma-Aldrich) and MilliQ water were used for RP-HPLC analyses and purification. Analytical RP-HPLC analyses were performed on a Dionex U3000SD with a Macherey-Nagel Nucleodur column (4.6 × 100 mm, 3 μm) at a flow rate of 1 mL min−1 at 50 °C. The mobile phase was composed of 0.1% (v/v) TFA–H2O (solvent A) and 0.1% TFA–CH3CN (solvent B). Purification was performed on a Gilson GX-281 with a Macherey-Nagel Nucleodur VP250/21 100-5 C18ec column (21 × 250 mm, 5 μm) at a flow rate of 20 mL min−1. LC-MS analyses were recorded in positive ion-mode with an ESI ion source on an Agilent™ Time-of-Flight MS mass spectrometer Model 6230 coupled with Agilent™ 1290 series front-end.Pyrrhocoricin (Py) was purchased at Novopro (Shanghai, China).
Peptides Onc112, Py and FITC-Py were synthesized on a Rink Amide PS resin with low loading (0.32 mmol g−1). Peptides Onc112-P7, Onc112-P7Scr, Py-P7, Py-P7Scr, FITC-Py-P7 and FITC-Py-P7Scr were synthesized using a Fmoc-Phe-Wang resin with low loading (0.31 mmol g−1) or Wang resin with low loading (0.30 mmol g−1). The attachment of the First Amino Acid on Wang Resin was performed from symmetrical anhydride method. 10.0 eq. of Fmoc-amino acid (relative to resin loading) were dissolved in dry DCM with a minimum amount of DMF. The reaction mixture was cooled to 0 °C and 5.0 eq. of DIC (relative to resin loading) were slowly added to the amino acid solution. The mixture was stirred for 20 min at 0 °C and DCM was evaporated. The residue was dissolved in a minimum of DMF and added to the resin suspension followed by 0.1 eq. of DMAP (relative to resin loading). The suspension is shaken at room temperature for 2 h.
HPLC and LCMS analyses of all peptides synthesized are presented in SI.1a (ESI†). HRMS analyses of all the fusion peptides synthesized are presented in SI.1b (ESI†).
Biological material
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 rpm) for 30 min at 4 °C. The supernatant was further centrifuged (47000 rpm) at 4 °C for 4 hours and the pellet resuspended in 12.5 mL of buffer A. This solution was laid on a 30% sucrose cushion and centrifuged (34000 rpm) for 19 hours at 4 °C. The ribosome pellet was resuspended in 12.5 mL buffer B (20 mM Tris HCl pH 7.5; 50 mM NH4Cl; 10 mM MgCl2; 0.1 mM EDTA; 6 mM β-mercaptoethanol) and centrifuged again (18200 rpm) for 1 hour at 4 °C. The supernatant was concentrated on Centrikon 100K (Millipore) and washed extensively with ITC 70S buffer. Ribosome concentration was determined by UV absorption at 260 nm. Samples were snap frozen in liquid nitrogen and kept at −80 °C.
200 rpm) for 30 min at 4 °C. The supernatant was further centrifuged (47000 rpm) at 4 °C for 4 hours and the pellet resuspended in 12.5 mL of buffer A. This solution was laid on a 30% sucrose cushion and centrifuged (34000 rpm) for 19 hours at 4 °C. The ribosome pellet was resuspended in 12.5 mL buffer B (20 mM Tris HCl pH 7.5; 50 mM NH4Cl; 10 mM MgCl2; 0.1 mM EDTA; 6 mM β-mercaptoethanol) and centrifuged again (18200 rpm) for 1 hour at 4 °C. The supernatant was concentrated on Centrikon 100K (Millipore) and washed extensively with ITC 70S buffer. Ribosome concentration was determined by UV absorption at 260 nm. Samples were snap frozen in liquid nitrogen and kept at −80 °C.
Isothermal titration calorimetry (ITC) experiments
ITC experiments were performed on an iTC200 (Microcal Malvern Panalytical) or PEAQ-ITC instrument (Microcal Malvern Panalytical). The E. coli SC were washed extensively with ITC buffer (R1 buffer: 10 mM Hepes pH 7.4, 0.15 M NaCl, 3 mM EDTA) on Centrikon 10K at 4 °C before ITC experiments. DLS analysis shows that the protein solution is monodisperse (data not shown). Similarly, E. coli 70S ribosome particles were also washed extensively with ITC 70S buffer (50 mM Tris HCl pH 7.5, 30 mM KCl, 70 mM NH4Cl, 7 mM MgCl2, 1 mM DTT, 5% glycerol) on Centrikon 100K at 4 °C before ITC experiments. Peptides (100 to 400 μM) were titrated at 30 °C (303.15 K) in sequential injections (usually 2 μl each) into a SC (20 or 30 μM) or 70S (10 to 15 μM) solution. Data were corrected from control experiments in which peptides were injected in ITC buffer solution. Each titration was performed at least twice. Analyses of experimental data were performed with AFFINImeter software (https://www.affinimeter.com; S4S, Santiago de Compostela, Spain). Thermodynamic profiles of the interactions and typical titration curves are presented in SI.2 and SI.3 (ESI†), respectively. All thermodynamic data and analysis are provided in SI.4 (ESI†).Peptides penetration assays
We analyzed the cell penetration capacity of the peptides in E. coli D22 cells on the basis of previously published work.16 FITC-labeled peptides were added to 50 μl of bacterial cultures (OD600 = 1) at a final concentration of 9 μM. The cells were allowed to acquire the peptides for 1 h at 37 °C in the dark and then washed with two volumes of PBS and finally resuspended in 10 μl of PBS. 1 μl of DAPI (final concentration: 15 μg mL−1) and 1 μl of Nile Red (final concentration: 80 μM) were added 15 minutes before spreading 2 μl of the stained cells on a thin 1.5% agarose pad poured on a microscope slide and finally covered with a coverslip. Cells were observed using a LSM780 inverted confocal microscope (Zeiss) equipped with a Plan Apo 63×/1.4 oil DIC M27 objective and using the 405, 488 and 514 nm excitation lasers and suitable filter sets for imaging the DAPI, FITC and Nile Red fluorescent signals, respectively. Images were processed using the open source Fiji software.27For the quantification of the relative peptide cell penetration capacity, cells were prepared for observation as above except that Nile Red staining was omitted. Cells were visualized using a Leica DM5500 microscope equipped with an HCX PL APO CS 63.0 × 1.40 Oil objective and using the filter systems L5 (488 nm) and A4 UV (365 nm) for detection of the FITC and DAPI signals, respectively. The relative peptide cell penetration capacity was performed, for each peptide and after background subtraction, by dividing the FITC signal by the one measured for the DAPI staining. Four sets of images for each peptide, representing a total of more than two hundred cells per peptide, were used to calculate the averaged relative signal of FITC and standard deviations measured for the four sets of images. Values presented in Fig. 2 were normalized to the one obtained with FITC-Py peptide, arbitrarily set equal to 100.
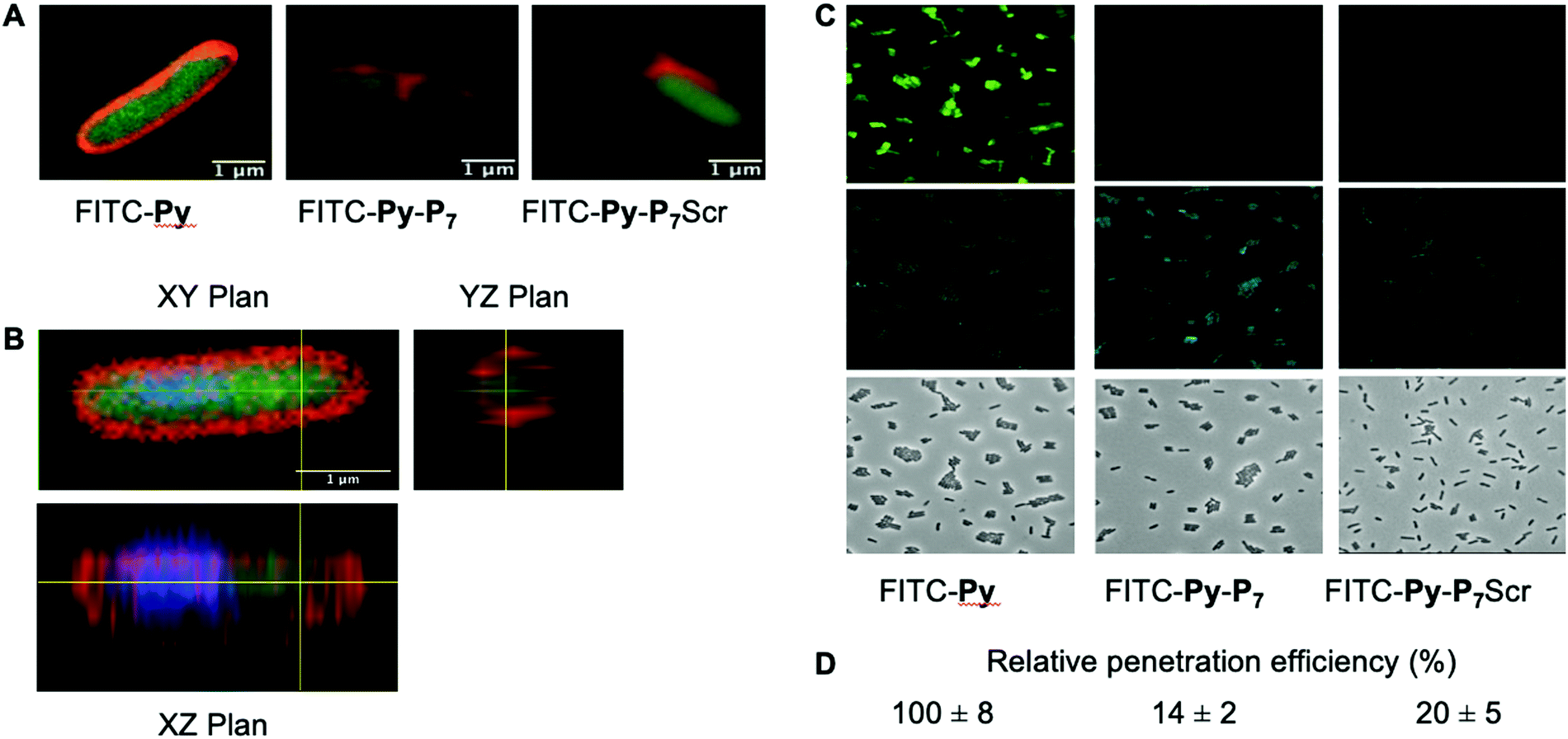 | ||
| Fig. 2 Analysis of peptide penetration into E. coli cells. (A) Confocal LSM of D22 E. coli cells exposed to the three different FITC labeled peptides (9 μM) and stained with Nile Red. Composite image were obtained by merging the FITC and Nile Red channels and clearly show the cytoplasmic localization of FITC. (B) Three-dimensional analysis of the FITC-Py (9 μM) peptide penetration in the bacterial cytoplasm. D22 cells were treated as in (A), except that they were additionally stained with DAPI (blue). It clearly appears that the two fluorescent signals from DAPI (blue) and FITC (green) are located within the cell, while Nile Red stains the lipidic phases of membranes. This unambiguously indicates the cytosolic localization of the peptide. (C) Representative fluorescent microscopy images of D22 E. coli cells exposed to the three different FITC labeled peptides (9 μM) and stained with DAPI. The uptake of the bifunctional peptides is clearly less efficient than the one of FITC-Py. (D) Relative FITC fluorescence quantification (average ± SD) normalized to that measured with FITC-Py peptide. Methods and quantification protocol are detailed in the Experimental section. | ||
Microdilution broth assays
E. coli D22 strains were cultivated on either agar or liquid 1× Mueller Hinton broth unless specified. MIC assays were performed in duplicate in 96-well microtiter plates on the basis of the protocol recommended by the Clinical and Laboratory Standards Institute. Briefly, a mix of 5–6 fresh (less than 36 hours) colonies grown on MH agar plates were inoculated in 3 mL of MH broth and incubated for about 16 h at 37 °C under agitation. 30 μl of this saturated culture were inoculated in 3 mL of fresh medium and allowed to grow until OD600 nm of about 0.6–0.8. Dilution to an OD600 nm of 0.002 was made in MH broth. Within a time laps not exceeding 30 min, 50 μl of this cell suspension was added to 50 μl of MH broth (containing or not the peptide/antibiotic diluted in MH broth) in wells of a 96-well round bottom microwell plate (Greiner; final cell density about 2 to 5 × 105 cfu per mL). Plates were incubated at 37 °C for 18–24 h and final turbidity of each well was measured at 620 nm using a Multiskan FC microplate reader (Thermo Scientific). MIC was defined as the minimal antibiotic concentration that result in no increase of turbidity at that time. Each MIC determination was performed in duplicate and at least in two independent experiments. Plating of 75 μl of the content of the wells of interest on LB Agar plates was used to determine the MBC that is defined by a decrease of at least 99.9% of cell viability as compared to the number of viable cells in the inoculum (about 2 to 5 × 104 cfu).Co-immunoprecipitation assays
1 U. OD600 of E. coli D22 cells freshly grown in MH broth were exposed for 1 hour in the dark and at 37 °C to H2O (no peptide) or to 16 μM of either FITC-Py, FITC-Py-P7Scr or FITC-Py-P7 peptide. After 5 min centrifugation at 6000 rpm and wash with 500 μl PBS, cells pellet was resuspended in 250 μl of lysis buffer A (50 mM Tris–HCl pH 7.6, 150 mM NaCl, 3 mM MgCl2, 1× cOmplete™ mini EDTA-free protease inhibitor cocktail (Roche)) and transferred to ice. Lysozyme was added at 1 mg mL−1 and incubation was prolonged for 30 min on ice. Suspensions were further sonicated (3 × 5 s plus 15 s cooling period) and Benzonase (0.5 μl of High Concentration solution, Merck) was added for an additional 30 min incubation on ice. Cell debris were removed by centrifugation at 14000g at 4 °C for 10 min. The supernatants were pre-cleared by incubation for 1 hour at 4 °C with 15 μl of 1/1 (v/v) protein G slurry (protein G Sepharose 4 Fast flow, GE Healthcare) previously washed and equilibrated with Buffer A. After centrifugation at 6000 rpm (4 °C, 5 min), 12.5 μl of the supernatants were used as input controls (input, lanes 1–4, Fig. 3 and SI.5, ESI†). To 220 μl of the lysates previously exposed to the FITC-labelled peptides (lanes 6–8, Fig. 3 and SI.5, ESI†), 4 μl of anti-FITC antibody (ab 19224, 1 mg mL−1, Abcam) was added and incubated for 80 min at 4 °C on a rotation wheel. The same protocol was applied to the sample exposed to H2O (lane 5, Fig. 3 and SI.5, ESI†), but without addition of the ab 19224 antibody. After 2 min centrifugation at 6000 rpm, the pellet was washed once with 250 μl and once with 100 μl of buffer A before final resuspension of the material in 10 μl of 5× SDS loading buffer. After 5 min heating at 95 °C, proteins were separated by a 10% SDS PAGE, transferred onto a PVDF membrane, stained with the Revert™ 700 Total Protein Stain (Li-Cor) and imaged on an Odyssey (Li-Cor) scanner according to the manufacturer protocol (SI.5A, ESI†). After a brief rinse with ddH2O, the membrane was further processed for the EcSC immunodetection using the mouse polyclonal anti-DnaN antibody (Ab246, C. McHenry, kindly provided by K. Marians; Fig. 3 and SI.5B, ESI†).
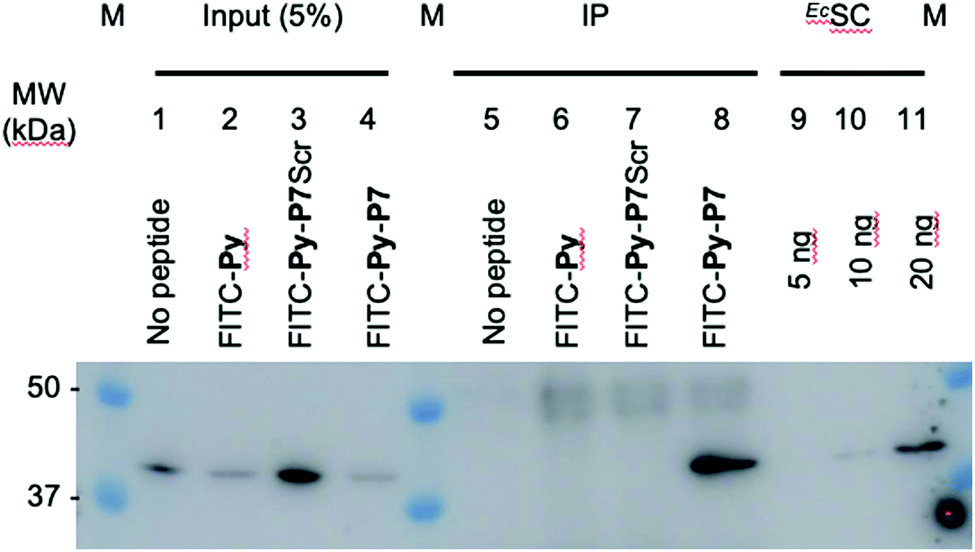 | ||
| Fig. 3 In cellulo targeting of the EcSC by the P7 peptide. E. coli D22 cells were grown in absence (no peptide) or presence of the indicated FITC-labelled peptides (16 μM) before extensive wash and lysis. Pull-down of the FITC-labelled peptides on the different pre-cleared lysates was carried out using anti-FITC antibody bound to protein G sepharose beads. Finally, the presence of the EcSC in the lysates (input) and in the pull-down fractions (ImmunoPrecipitated, IP) was assessed by western blotting using a polyclonal anti-DnaN antibody (Ab246, kindly provided by Charles McHenry and Kenneth Marians). The specificity and sensitivity of the anti-DnaN antibody are shown using whole cell extracts (input) and 5, 10 and 20 ng of purified EcSC (DnaN),8 respectively. Full gel image and total protein stain of this representative assay are shown in SI.5 (ESI†). The specific FITC-Py-P7 mediated co-immunoprecipitation of the SC was confirmed in 3 independent biological replicates. | ||
In vivo survival assay using a D. melanogaster infection model
50 mL of E. coli ATCC23754 were grown at 29 °C in LB media up to exponential phase (OD600 = 0.6–0.8) and centrifuged at 5.000g for 5 min. The pellet was then diluted in 1 mL of PBS. Immune-deficient kennyC028328 flies were raised at 25 °C with 60% humidity on standard cornmeal-agar medium. Seven-day-old female flies were infected by septic injury with a thin tungsten needle previously dipped in an E. coli suspension diluted in PBS and kept at 29 °C. One hour and 24 hours after infection, 18.4 nL of peptides (1 mM) diluted in PBS or PBS alone were injected into the fly body cavity (Nanoject II apparatus; Drummond Scientific). Each group enrolled 60 flies and the experiment was repeated 3 times independently. Log-rank analysis of each individual assay was performed with the OASIS online application.29 Because log-rank analysis can only compare two survival curves at the same time and during the same experiment, we compute the median lethal time 50 (LT50) and the survival rate at 144 hours of the 3 independent experiments to perform one-way ANOVA statistical analysis. To control the innocuity of the peptides and of the protocol, the experiments were repeated in the same conditions except the flies were injured with a sterile needle.
Results and discussion
In cellulo overexpression of the consensus SC binding motif demonstrates cytotoxic activity
We previously showed that the small peptides we have developed efficiently inhibit in vitro SC dependent DNA polymerization by bacterial polymerases.3,8 On the other hand, in cellulo overexpression of full-length SC binding proteins such as DNA PolIV or its catalytically dead mutant prevent cell growth by inhibiting DNA replication thanks to their SC binding motifs.30 However, the ability of a short SC binding peptide to inhibit cell growth was not yet demonstrated. We cloned such a short SC binding motif (8F: MGPRQLDLF) in fusion with GST in an IPTG inducible expression system. As controls, we used both the empty vector and a construct containing the SC binding motif mutated in three positions essential for the efficient binding to the SC (8A: MGPRALDAA). As shown in Fig. 1A, only the expression, under induced (+IPTG) conditions, of the bona fide SC binding peptide precludes the colony forming capacity of the transformed cells. Similarly, expression of the SC binding peptide but not the mutant form inhibits the growth of E. coli in liquid cultures (Fig. 1B). These experiments thus demonstrate the capacity of short SC binding peptides to inhibit cell growth of E. coli.Mono- and bifunctional peptides used in the study
To directly assess the antimicrobial activity of SC binding peptides on E. coli, we synthesized a series of mono- and bifunctional peptides (Table 1). Peptide P7 (AcQXDLF, X = cyclohexylalanyl, Cha) has been previously developed using a structure-guided approach,3 starting from the native SC binding sequence (RQLVLGL) from the E. coli DNA polymerase IV.6,10 The affinity of P7 for EcSC shows a 50-fold increase compared to the native sequence, as determined by SPR.3 Pyrrhocoricin (Py) is the non-glycosylated version of a natural PrAMP from Pyrrhocoris apterus31 and Onc112 is an optimized synthetic peptide derived from a natural oncocin from the Hemiptera Oncopeltus fasciatus, containing D-amino acids to increase its stability.24 Both peptides interact within the PEC of Gram-negative bacteria 70S ribosomes,21,32 block the peptidyl transferase center and ultimately inhibit translation.19,21,33 Another intracellular target of PrAMPs, the protein chaperone DnaK, was also identified but the PrAMP–DnaK interaction is not considered as the major molecular event responsible for bacterial growth inhibition.19 A series of bifunctional peptides joining covalently P7 and PrAMP sequences was prepared as well as fluorescently labelled (FITC) versions to track the penetration efficiency of the peptides into bacterial cells. Standard solid-phase peptide synthesis methods were employed and a short linker (N)GPR was introduced to connect the N-terminus of P7 to the C-terminus of PrAMP sequences. All peptides used in this study are presented in Table 1.Bifunctional peptides readily interact with both targets in vitro
The interactions of the different peptides with their respective targets (i.e.EcSC and Ec70S ribosome) were monitored by ITC at 30 °C and the resulting KD values are presented in Table 1. Full thermodynamic profiles, values, typical ITC titration curves and analysis for each type of interaction are shown in SI.2–4 (ESI†).As expected from previous studies, P7 efficiently interacts with EcSC3,23 (Table 1) whereas PrAMPs Onc112, Py and FITC-Py do not (SI.2B, E and H, ESI†). In contrast, the bifunctional peptides, resulting from the N-terminal conjugation of P7 to each PrAMP, i.e.Onc112-P7, Py-P7 and FITC-Py-P7 interact with EcSC as efficiently as P7 alone and present similar thermodynamic profiles (Table 1 and Fig. SI.2A, SI.3C, F, I, ESI†). Control peptide conjugates, Onc112-P7Scr, Py-P7Scr and FITC-Py-P7Scr were synthesized using a scrambled variant of P73 (P7Scr, XFQLD, X = cyclohexylalanyl, Cha). These peptides fail to interact with EcSC (Table 1 and SI.3D, G, J, ESI†), underlining the sequence specificity of the P7 interaction within the EcSC pocket.
The interaction of each peptide with the Ec70S ribosomal particle was also characterized by ITC (Table 1 and SI.2B, SI.3K–T, ESI†). P7 does not specifically bind to Ec70S but some unspecific binding might be observed (SI.3K, ESI†). In contrast, unlabeled PrAMPs interact with high affinity with their target (Table 1 and SI.2B, SI.3L, O, ESI†), with KD values of 84 and 15 nM for Onc112 and Py, respectively. The bifunctional peptides Onc112-P7, Py-P7 and the control version with scrambled P7 also interact with Ec70S with affinities similar to the PrAMPs alone, indicating that C-terminal extension of the PrAMPs with a SC-binding sequence does not compromise the interaction with the ribosomal target (Table 1 and Fig. SI.2B, SI.3M, P, Q, ESI†). Finally, the conjugation of FITC at the N-terminus of Py and related bifunctional peptides Py-P7 and Py-P7Scr was found to be deleterious for the interaction with Ec70S (Table 1 and SI.3R–T, ESI†). Similar thermodynamic profiles are observed for both types of peptides (SI.2B, ESI†): the spontaneous interaction is characterized by an exothermic component, in line with the formation of hydrogen bonds between peptides residues and 23S nucleotides,21,32 and a non favorable entropic factor partly accounting for the loss of freedom degree of the ligand upon binding (see additional discussion in SI.4, ESI†). To the best of our knowledge, the data presented in SI.2–4 (ESI†) are the first report of the thermodynamic analysis of PrAMPs–ribosome interactions.
In conclusion of these interaction studies, we show that the bifunctional peptides (Onc112-P7 and Py-P7) retain the binding characteristics of each cognate peptide (Table 1 and SI.2, ESI†), notably in terms of specificity. Indeed, peptide conjugates are able to bind both targets, namely EcSC and Ec70S, whereas their parental peptides are specific to only one. Finally, our experiments with FITC-labeled peptides reveal that the N-terminal added moiety drastically affects the peptide interaction with Ec70S, possibly by hindering its entry into the PEC. This last result is consistent with the crystal structures of PrAMPs in complex with the ribosome.32,34
PrAMPs and bifunctional peptides accumulate differently into E. coli cytoplasm
We then checked by confocal microscopy whether the synthetic peptides penetrate into the bacterial cytoplasm (Fig. 2), as previously reported,35 and do not solely accumulate at the membrane level or in the periplasm (Fig. 2A). The image analysis along the Z-axis (Fig. 2B) undoubtedly shows that the FITC fluorescence signal of the three fluorescently labelled peptides, namely FITC-Py, FITC-Py-P7 and FITC-Py-P7Scr colocalizes with DAPI, indicating that the peptides enter the bacterial cytoplasm delimited by the membrane-specific hydrophobic dye Nile Red36 (Fig. 2A and B). The accumulation of these peptides in bacteria was quantified (Fig. 2C and D). Comparison of the intensity of FITC staining of E. coli cells after exposure to equal concentrations of FITC-Py, FITC-Py-P7 or FITC-Py-P7Scr reveals that the fluorophore-tagged bifunctional peptides enter the cells about 5–7 times less efficiently than FITC-Py (Fig. 2C and D). We conclude that the additional C-terminal SC binding motif in bifunctional sequences reduces but does not suppress transport properties of the PrAMP within the bacteria.SC binding peptide demonstrate antibacterial activity against E. coli cells
We have demonstrated that expression of short SC binding peptide fused to GST within E. coli cells inhibits cell growth (Fig. 1). Knowing that P7 fused to PrAMPs are effectively transported into the bacterial cytoplasm (Fig. 2), we next determined the antibacterial potency (Minimal Inhibitory Concentration or MIC) of the synthesized compounds by the microdilution broth assay (Table 2). P7 alone fails to induce any detectable antibacterial activity. On the other hand, PrAMPs like Onc112 and Py demonstrate antibacterial activities in the micromolar range in agreement with previous reports.31,37,38 The differences in MIC values observed between these studies may be attributed to the use of different E. coli strains and/or culture media. The higher activity of Onc112 relative to Py, despite a lower affinity for the ribosome (Table 1), probably relies on its higher uptake by the E. coli cells and/or to its engineered increased stability24 but also to the potential engagement of additional targets. Minimal Bactericidal Concentration (MBC) determination for Py and Onc derived peptides confirms the bactericidal effect of these peptides, which appear to be 1 to 16 times more potent than ampicillin (Table 2). The observed MIC values do not reveal any significant difference in antibacterial activity for the different unlabeled bifunctional peptides except for Onc112-P7 that appears to be twice more active than Onc112-P7Scr (Table 2). We reasoned that this a priori disappointing result might be linked to the ratio of the different targets within the cell. Indeed, the relevant target of the PrAMPs for what concern toxicity, the 70S ribosome, is present at about 8000 to 73000 copies per cell, depending on the metabolic status, as compared to about 300 copies of dimeric SC.39,40 Given that the affinities of the two peptides Onc112-P7 and Py-P7 for the ribosome are three-fold higher than for the EcSC (Table 1), the binding equilibrium of the bifunctional peptides is theoretically largely displaced toward the binding to the most abundant target, i.e. the ribosome. This phenomenon might, in turn, mask the possibility to observe any substantial antibacterial effect that would result from EcSC engagement by the P7 component of bifunctional peptides. This reasoning may also explain why no clear decrease in antimicrobial activity is observed with peptides Onc112-P7Scr and Py-P7Scr, which both include a scrambled version of P7, as compared to their EcSC binding derivatives Onc112-P7 and Py-P7 (Table 2). According to this hypothesis, eliminating or lowering the affinity of the bifunctional peptides toward the ribosome may allow the antibacterial activity of P7 to be revealed. To this end, we took advantage of the N-terminal FITC labeling of the Py containing peptides that we showed to be deleterious to their ribosome binding capacities (Table 1 and SI.3R–T, ESI†). We assessed the MIC values for each of these N-ter FITC modified peptides (Table 2). The strong decrease in antibacterial efficiency of FITC-Py as compared to Py (64 to 512 fold increase in MIC; Table 2) is consistent with the reduced affinity for its ribosomal target measured by ITC (Table 1), and is a strong argument in favor of a predominant PrAMP/70S interaction in vivo. In combination with microscopy analysis, these data confirm that the FITC moiety blocks the interaction with the ribosome but does not preclude the peptide transport into the cell (Fig. 2). The residual antibacterial activity of FITC-Py peptide may be due to binding to the DnaK chaperone as described previously41 or to other intracellular targets. Upon fusing P7 to FITC-Py, resulting in FITC-Py-P7, and despite a 7-fold lower cell penetration capacity (Fig. 2C and D), the MIC value drops by a factor 4 to 16 (Table 2). This reveals a specific toxic effect of P7 and is further strengthened by the observation that a scrambled P7 sequence fused to FITC-Py (FITC-Py-P7Scr) fails to show a similar toxic effect (Table 2).
| Compound | MICa | MBCb | |
|---|---|---|---|
| μM | μg mL−1 | μg mL−1 | |
| Ampicillin and tetracycline antibiotics were included in the assays as positive controls. n.d.: not determined.a Ranges of minimal inhibitory concentrations determined in a minimum of 2 independent biological replicates are indicated.b Minimal bactericidal concentrations determined in a minimum of 2 independent biological replicates are indicated. See the Experimental section for detailed procedures. | |||
| P 7 | >357 | >256 | n.d. |
| Onc112 | 0.21–0.42 | 0.5–1 | 1 |
| Onc112-P 7 | 0.15–0.29 | 0.5–1 | 2 |
| Onc112-P 7 Scr | 0.29–0.58 | 1–2 | 2 |
| Py | 0.85–3.4 | 2–8 | 8 |
| Py-P 7 | 1.2–2.4 | 4–8 | 8 |
| Py-P 7 Scr | 1.2–4.8 | 4–16 | 16 |
| FITC-Py | 181–362 | 512–1024 | n.d. |
| FITC-Py-P7 | 16.9–33.8 | 64–128 | 128 |
| FITC-Py-P7Scr | >269.9 | >1024 | n.d. |
| Ampicillin | 22.9–45.8 | 8–16 | 16 |
| Tetracycline | 0.56–1.12 | 0.25–0.5 | 0.5 |
To further consolidate the specific effect of the SC binding peptide, we performed immunoprecipitation of the FITC-labelled peptides after incubation with E. coli cells (Fig. 3). As shown, only the bona fide SC interacting peptide (i.e. FITC-Py-P7, lane 8) but not the FITC-Py (lane 6) or the FITC-Py-P7Scr (lane 7) can efficiently mediate the co-immunoprecipitation of the EcSC (Fig. 3 and SI.5, ESI†). Altogether, these data unambiguously reveal a specific P7-dependent bactericidal effect that can be attributed to the engagement of EcSC, according to our hypothesis and our previous in vitro works.3,8
The Onc112-P7 bifunctional peptide displays a synergic protective activity in a Drosophila melanogaster infection assay
In order to further investigate the validity of our approach, we tested the efficiency of the most potent bifunctional peptide, as measured in vitro (i.e.Onc112-P7, Table 2), to control an in vivo infection using E. coli sensitive D. melanogaster mutant flies.The protocol assay in absence of infection has no effect on flies’ survival (Fig. 4A) showing the non-toxicity of the different peptides in this system at the specified doses used. We then performed three independent sets of experiments which are highly reproducible (SI.6, ESI†). For each independent experiment, a log rank analysis reveals a clear and statistically significant effect of peptides over PBS on flies’ survival. This statistic analysis does not display such a significant effect when comparing the different peptides in the first two days because the curves are very similar. However, an increased activity of the Onc112-P7 peptide, as compared to the two others, is clearly observed after about 60 hours (SI.6, ESI†). We pooled the data of these three independent experiments (Fig. 4B) and derived the survival at 144 h after infection and the lethal time 50% (LT50) (Fig. 4C and D, respectively).
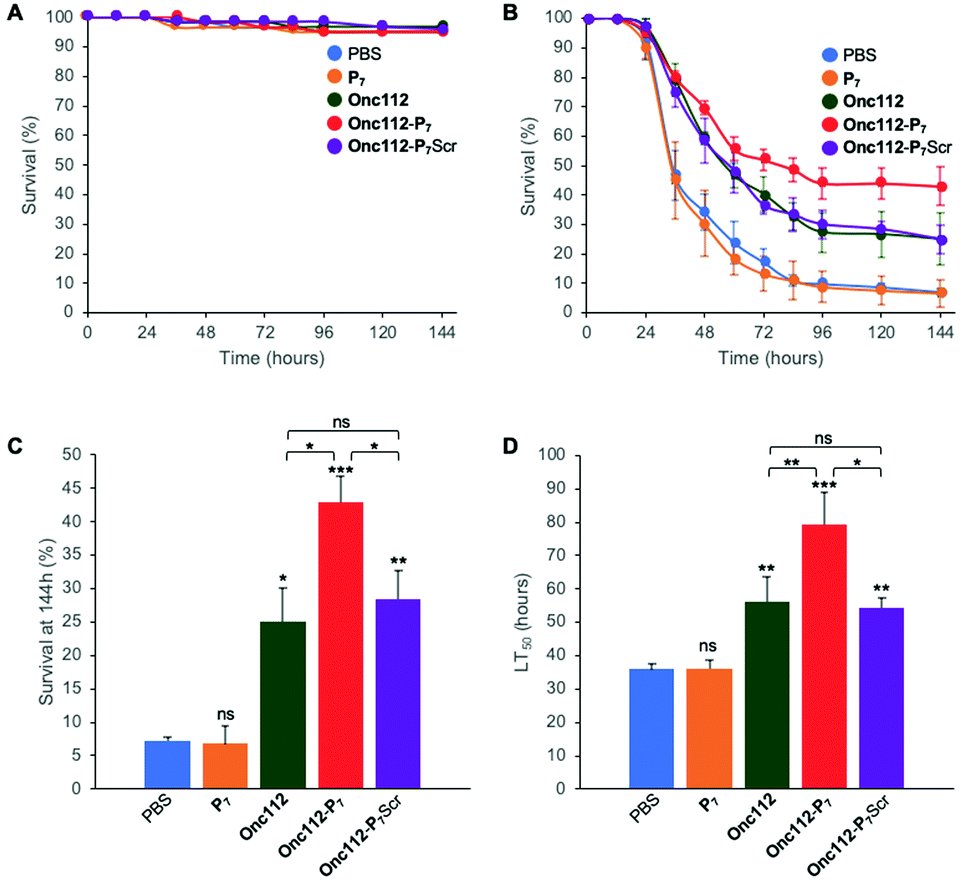 | ||
| Fig. 4 In vivo survival assay using Drosophila melanogaster infection model. Seven-day-old female kennyC02831 flies were subjected to sterile injury (A) or septic injury (B) with a thin tungsten needle previously dipped in sterile PBS alone or in an E. coli suspension diluted in PBS and kept at 29 °C. 1 and 24 hours after injury, 18.4 nL of peptides (1 mM) diluted in PBS or PBS alone were injected into the flies body cavity. Data represents means ± standard errors of 3 independent experiments. Individual experiments and their respective log-rank analysis are presented in SI.5 (ESI†). The survival rate at 144 hours (C) and the Lethal Time 50% (LT50) (D) represent the mean ± standard errors of the value determined for each of the 3 independent experiments and data were analyzed by the One-Way ANOVA test: *P < 0.05, **P < 0.01, ***P < 0.001. ns: not significant. | ||
P 7 peptide has no effect on fly survival (Fig. 4B–D), in agreement with our previous observations that it does not have any antibacterial activity on E. coli cultures (Table 2). Onc112 has a protective effect against E. coli infection as it induces a 3-fold increased survival of infected flies 144 h after injection (Fig. 4B–D). This is in line with the known in vivo antimicrobial activity of PrAMPs in murine infection models.42,43
Remarkably, Onc112-P7 increases the flies’ survival by a factor 4.5 whereas Onc112-P7Scr does not improve survival as compared to Onc112. This is indicative of an improved effect of the bifunctional peptide as compared to the cumulative effects of P7 and Onc112. This synergistic effect conceptually results from the combination of both the translation inhibition by the PrAMP component (Onc112) and the SC binding related toxicity of P7 (Fig. 4B and C). This is also clearly observed when considering the LT50 curve (Fig. 4D) showing that Onc112-P7 substantially increases the flies’ lifetime (i.e. by about 40%) as compared to Onc112 and Onc112-P7Scr. Consequently, it appears that the biological effect of Onc112-P7 is substantially different from that of Onc112 and Onc112-P7Scr, presumably because of its bifunctional nature.
Given the only modest increase in in vitro toxicity observed for Onc112-P7 as compared to Onc112 and Onc112-P7Scr (Table 2), these in vivo results show that some other parameters may modulate the antibacterial activity of the peptides in vivo. For example, the modification of the peptidic sequence may change the pharmacokinetics properties of either peptide or other physiological parameters such as pH may alter the output of the experiment.44,45 Alternatively, in line with the observed increased antibacterial activity of FITC-Py-P7 as compared to FITC-Py and FITC-Py-P7Scr (Table 2), the P7 mediated protective effect observed on flies survival might result from a lower availability of the bacterial ribosomal target for the bifunctional Onc112-P7 peptide which, as a consequence, is partly redirected towards the bacterial SC. Differences in metabolic activity of the bacterial cells, between in vitro (unstressed) and in vivo (stressful) situations, may greatly influence the numbers of ribosomal targets39 and differences in target accessibility may also depend on the translating status/activity of the cell, thus potentially reducing the contribution of the PrAMP moiety. Another argument to explain the better in vivo activity of the bifunctional Onc112-P7 peptide, as compared to in vitro situation, may rely on the endogenous production of PrAMPs and others Drosophila Immune induced Molecules (DIMs) in the flies’ hemolymph upon bacterial infection, even in immune deficient (imd) flies,46 that can modulate the integrated biological effect of the peptides by different ways (number of available targets, membrane integrity,…).
Conclusions
The data presented in this paper demonstrate that, in E. coli, peptides specifically designed to block the SC-DNA polymerases interaction and that were shown to inhibit the replicase activity in vitro3,47 readily induce bacterial cell death, whether they are produced from within the cells or imported via a transfecting peptide. Moreover, this bactericidal activity leads to a protective effect in living animals challenged by a bacterial infection. We previously determined the high degree of conservation of key residues involved in the binding of short peptides within the SC binding pocket among 128 bacteria from various phylae.8 Moreover, there is a strict conservation of the SC binding pocket between E. coli and other human pathogens such as K. pneumoniae, P. aeruginosa and A. Baumanii (among others), all identified by OMS as critical priority strains. Collectively, these results further validate SC as a molecular target for the development of a new class of antibiotics. To the best of our knowledge, this is the first demonstration of an in vitro and in vivo antibacterial activity of a SC binding molecule issued from a structure-based approach against Gram-negative bacteria. Other works have screened molecules libraries in search for SC inhibitors with antibacterial activities48 or demonstrated that the anti-tuberculosis activity of already known drugs such as streptomyces-derived griselimycin, is mediated through the binding to SC.49 These studies identified active molecules that are, at best, 2 to 70-fold less efficient on E. coli than the FITC-Py-P7 compound described here. Remarkably, Kling and coworkers49 showed that resistance of Mycobacterium smegmatis to griselimycin occurred at the extremely low frequency of 5 × 10−10 highlighting thus the potential of SC binding molecules to reduce the rising of antimicrobial resistance.Our study also demonstrates that the use of bifunctional antimicrobial agents, selective for two different cellular targets involved in distinct and essential metabolic pathways, may trigger a synergistic effect. By its involvement in DNA replication and mutagenesis and its yet unexploited potential as antimicrobial target, the bacterial SC represents a chance to develop antibiotics that will contribute to the fight against antimicrobial resistance.9,50 Moreover, targeting simultaneously two distinct and essential metabolic pathways mathematically reduce the probability of resistance acquisition. The developments of bifunctional peptides with increased affinity for SC, improved stability and bioavailability, bacterial cell delivery and efficiency in arresting these two major biochemical pathways are in progress.
Author contribution
D. B., G. G. and J. E. W. conceived the project. C. N. helped to develop the methodological framework. C. A., G. C. and C. M. designed and performed the peptides synthesis and analyses. P. W., A.-M. L. and D. B. purified the biological material, designed and performed the ITC experiments. J. E. W. designed and performed the in cellulo overexpression assays, the microdilution assays, the peptide penetration assays and the co-immunoprecipitation assays. J.-M. R. and F. V. designed, performed and analyzed the D. melanogaster infection assays. D. B., G. G., J.-M. R., F. V. and J. E. W. analyzed the data. D. B., G. G. and J. E. W. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by a pre-maturation program from SATT Alsace Conectus. A doctoral fellowship from DGA and Conseil Régional de Nouvelle Aquitaine (to C. M.) is gratefully acknowledged. We deeply thank Charles McHenry and Kenneth Marians for the kind gift of the anti DnaN antibodies. We thank Cyrielle Silva da Vega and Karl Brillet for their help in ITC experiments. We thank Dr Philippe Bulet for constructive discussions, J. Mutterer for his help with confocal microscopy experiments, and Drs J. C. Amé and G. Reina for their help with ImageJ.References
- Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics, https://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/en/, (accessed April 2020).
- X. P. Kong, R. Onrust, M. O’Donnell and J. Kuriyan, Three-dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp, Cell, 1992, 69, 425–437 CrossRef CAS PubMed.
- P. Wolff, V. Oliéric, J. P. Briand, O. Chaloin, A. Dejaegere, P. Dumas, E. Ennifar, G. Guichard, J. Wagner and D. Y. Burnouf, Structure-Based Design of Short Peptide Ligands Binding onto the E. coli Processivity Ring, J. Med. Chem., 2011, 54, 4627–4637 CrossRef CAS PubMed.
- A. S. Altieri and Z. Kelman, DNA Sliding Clamps as Therapeutic Targets, Front. Mol. Biosci., 2018, 5, 87 CrossRef CAS PubMed.
- O. J. Becherel, R. P. Fuchs and J. Wagner, Pivotal role of the β-clamp in translesion DNA synthesis and mutagenesis in E. coli cells, DNA Repair, 2002, 1, 703–708 CrossRef CAS PubMed.
- B. P. Dalrymple, K. Kongsuwan, G. Wijffels, N. E. Dixon and P. A. Jennings, A universal protein–protein interaction motif in the eubacterial DNA replication and repair systems, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11627–11632 CrossRef CAS PubMed.
- K. A. Bunting, S. M. Roe and L. H. Pearl, Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the beta-clamp, EMBO J., 2003, 22, 5883–5892 CrossRef CAS PubMed.
- D. Y. Burnouf, V. Olieric, J. Wagner, S. Fujii, J. Reinbolt, R. P. P. Fuchs and P. Dumas, Structural and Biochemical Analysis of Sliding Clamp/Ligand Interactions Suggest a Competition Between Replicative and Translesion DNA Polymerases, J. Mol. Biol., 2004, 335, 1187–1197 CrossRef CAS PubMed.
- R. T. Cirz, J. K. Chin, D. R. Andes, V. de Crécy-Lagard, W. A. Craig and F. E. Romesberg, Inhibition of mutation and combating the evolution of antibiotic resistance, PLoS Biol., 2005, 3, e176 CrossRef PubMed.
- N. Lenne-Samuel, J. Wagner, H. Etienne and R. P. Fuchs, The processivity factor β controls DNA polymerase IV traffic during spontaneous mutagenesis and translesion synthesis in vivo, EMBO Rep., 2002, 3, 45–49 CrossRef CAS PubMed.
- L. Otvos, Antibacterial peptides isolated from insects, J. Pept. Sci., 2000, 6, 497–511 CrossRef CAS PubMed.
- M. Graf, M. Mardirossian, F. Nguyen, A. C. Seefeldt, G. Guichard, M. Scocchi, C. A. Innis and D. N. Wilson, Proline-rich antimicrobial peptides targeting protein synthesis, Nat. Prod. Rep., 2017, 34, 702–711 RSC.
- M. Mattiuzzo, A. Bandiera, R. Gennaro, M. Benincasa, S. Pacor, N. Antcheva and M. Scocchi, Role of the Escherichia coli SbmA in the antimicrobial activity of proline-rich peptides, Mol. Microbiol., 2007, 66, 151–163 CrossRef CAS PubMed.
- A. Krizsan, D. Knappe and R. Hoffmann, Influence of the yjiL-mdtM Gene Cluster on the Antibacterial Activity of Proline-Rich Antimicrobial Peptides Overcoming Escherichia coli Resistance Induced by the Missing SbmA Transporter System, Antimicrob. Agents Chemother., 2015, 59, 5992–5998 CrossRef CAS PubMed.
- K. Sadler, K. D. Eom, J.-L. Yang, Y. Dimitrova and J. P. Tam, Translocating proline-rich peptides from the antimicrobial peptide bactenecin 7, Biochemistry, 2002, 41, 14150–14157 CrossRef CAS PubMed.
- L. Otvos, M. Cudic, B. Y. Chua, G. Deliyannis and D. C. Jackson, An insect antibacterial peptide-based drug delivery system, Mol. Pharmaceutics, 2004, 1, 220–232 CrossRef CAS PubMed.
- W. Tian, B. Li, X. Zhang, W. Dang, X. Wang, H. Tang, L. Wang, H. Cao and T. Chen, Suppression of tumor invasion and migration in breast cancer cells following delivery of siRNA against Stat3 with the antimicrobial peptide PR39, Oncol. Rep., 2012, 28, 1362–1368 CrossRef CAS PubMed.
- M. Mardirossian, R. Sola, B. Beckert, D. W. P. Collis, A. Di Stasi, F. Armas, K. Hilpert, D. N. Wilson and M. Scocchi, Proline-Rich Peptides with Improved Antimicrobial Activity against E. coli, K. pneumoniae, and A. baumannii, ChemMedChem, 2019, 14, 2025–2033 CrossRef CAS PubMed.
- A. Krizsan, D. Volke, S. Weinert, N. Sträter, D. Knappe and R. Hoffmann, Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70S ribosome, Angew. Chem., Int. Ed., 2014, 53, 12236–12239 CrossRef CAS PubMed.
- D. Volke, A. Krizsan, N. Berthold, D. Knappe and R. Hoffmann, Identification of Api88 Binding Partners in Escherichia coli Using a Photoaffinity-Cross-Link Strategy and Label-Free Quantification, J. Proteome Res., 2015, 14, 3274–3283 CrossRef CAS PubMed.
- A. C. Seefeldt, F. Nguyen, S. Antunes, N. Pérébaskine, M. Graf, S. Arenz, K. K. Inampudi, C. Douat, G. Guichard, D. N. Wilson and C. A. Innis, The proline-rich antimicrobial peptide Onc112 inhibits translation by blocking and destabilizing the initiation complex, Nat. Struct. Mol. Biol., 2015, 22, 470–475 CrossRef CAS PubMed.
- P. Wolff, I. Amal, V. Oliéric, O. Chaloin, G. Gygli, E. Ennifar, B. Lorber, G. Guichard, J. Wagner, A. Dejaegere and D. Y. Burnouf, Differential Modes of Peptide Binding onto Replicative Sliding Clamps from Various Bacterial Origins, J. Med. Chem., 2014, 57, 7565–7576 CrossRef CAS PubMed.
- C. André, I. Martiel, P. Wolff, M. Landolfo, B. Lorber, C. Silva da Veiga, A. Dejaegere, P. Dumas, G. Guichard, V. Oliéric, J. Wagner and D. Y. Burnouf, Interaction of a Model Peptide on Gram negative and Gram positive Bacterial Sliding Clamps, ACS Infect. Dis., 2019, 5(6), 1022–1034, DOI:10.1021/acsinfecdis.9b00089.
- D. Knappe, N. Kabankov and R. Hoffmann, Bactericidal oncocin derivatives with superior serum stabilities, Int. J. Antimicrob. Agents, 2011, 37, 166–170 CrossRef CAS PubMed.
- R. Hoffmann, P. Bulet, L. Urge and L. Otvös, Range of activity and metabolic stability of synthetic antibacterial glycopeptides from insects, Biochim. Biophys. Acta, 1999, 1426, 459–467 CrossRef CAS.
- A. Dümmler, A.-M. Lawrence and A. de Marco, Simplified screening for the detection of soluble fusion constructs expressed in E. coli using a modular set of vectors, Microb. Cell Fact., 2005, 4, 1–10 CrossRef PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Fiji: an open-source platform for biological-image analysis, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- R. Bou Aoun, C. Hetru, L. Troxler, D. Doucet, D. Ferrandon and N. Matt, Analysis of Thioester-Containing Proteins during the Innate Immune Response of Drosophila melanogaster, J. Innate Immun., 2010, 3, 52–64 CrossRef PubMed.
- J.-S. Yang, H.-J. Nam, M. Seo, S. K. Han, Y. Choi, H. G. Nam, S.-J. Lee and S. Kim, OASIS: Online Application for the Survival Analysis of Lifespan Assays Performed in Aging Research, PLoS One, 2011, 6, e23525 CrossRef CAS PubMed.
- K. Uchida, A. Furukohri, Y. Shinozaki, T. Mori, D. Ogawara, S. Kanaya, T. Nohmi, H. Maki and M. Akiyama, Overproduction of Escherichia coli DNA polymerase DinB (Pol IV) inhibits replication fork progression and is lethal, Mol. Microbiol., 2008, 70, 608–622 CrossRef CAS PubMed.
- S. Cociancich, A. Dupont, G. Hegy, R. Lanot, F. Holder, C. Hetru, J. A. Hoffmann and P. Bulet, Novel inducible antibacterial peptides from a hemipteran insect, the sap-sucking bug Pyrrhocoris apterus, Biochem. J., 1994, 300(Pt 2), 567–575 CrossRef CAS PubMed.
- A. C. Seefeldt, M. Graf, N. Pérébaskine, F. Nguyen, S. Arenz, M. Mardirossian, M. Scocchi, D. N. Wilson and C. A. Innis, Structure of the mammalian antimicrobial peptide Bac7(1-16) bound within the exit tunnel of a bacterial ribosome, Nucleic Acids Res., 2016, 44, 2429–2438 CrossRef CAS PubMed.
- M. Taniguchi, A. Ochiai, H. Kondo, S. Fukuda, Y. Ishiyama, E. Saitoh, T. Kato and T. Tanaka, Pyrrhocoricin, a proline-rich antimicrobial peptide derived from insect, inhibits the translation process in the cell-free Escherichia coli protein synthesis system, J. Biosci. Bioeng., 2016, 121, 591–598 CrossRef CAS PubMed.
- M. G. Gagnon, R. N. Roy, I. B. Lomakin, T. Florin, A. S. Mankin and T. A. Steitz, Structures of proline-rich peptides bound to the ribosome reveal a common mechanism of protein synthesis inhibition, Nucleic Acids Res., 2016, 44, 2439–2450 CrossRef CAS PubMed.
- G. Kragol, R. Hoffmann, M. A. Chattergoon, S. Lovas, M. Cudic, P. Bulet, B. A. Condie, K. J. Rosengren, L. J. Montaner and L. Otvos, Identification of crucial residues for the antibacterial activity of the proline-rich peptide, pyrrhocoricin, Eur. J. Biochem., 2002, 269, 4226–4237 CrossRef CAS PubMed.
- C. K. Spahn, M. Glaesmann, J. B. Grimm, A. X. Ayala, L. D. Lavis and M. Heilemann, A toolbox for multiplexed super-resolution imaging of the E. coli nucleoid and membrane using novel PAINT labels, Sci. Rep., 2018, 8, 14768 CrossRef PubMed.
- L. Holfeld, R. Hoffmann and D. Knappe, Correlating uptake and activity of proline-rich antimicrobial peptides in Escherichia coli, Anal. Bioanal. Chem., 2017, 409, 5581–5592 CrossRef CAS PubMed.
- A. Hansen, I. Schäfer, D. Knappe, P. Seibel and R. Hoffmann, Intracellular toxicity of proline-rich antimicrobial peptides shuttled into mammalian cells by the cell-penetrating peptide penetratin, Antimicrob. Agents Chemother., 2012, 56, 5194–5201 CrossRef CAS PubMed.
- H. Bremer and P. P. Dennis, Modulation of Chemical Composition and Other Parameters of the Cell at Different Exponential Growth Rates, EcoSal Plus, 2008, 3(1) DOI:10.1128/ecosal.5.2.3.
- P. M. Burgers, A. Kornberg and Y. Sakakibara, The dnaN gene codes for the beta subunit of DNA polymerase III holoenzyme of Escherichia coli, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 5391–5395 CrossRef CAS PubMed.
- G. Kragol, S. Lovas, G. Varadi, B. A. Condie, R. Hoffmann and L. Otvos, The Antibacterial Peptide Pyrrhocoricin Inhibits the ATPase Actions of DnaK and Prevents Chaperone-Assisted Protein Folding, Biochemistry, 2001, 40, 3016–3026 CrossRef CAS PubMed.
- P. Czihal, D. Knappe, S. Fritsche, M. Zahn, N. Berthold, S. Piantavigna, U. Müller, S. Van Dorpe, N. Herth, A. Binas, G. Köhler, B. De Spiegeleer, L. L. Martin, O. Nolte, N. Sträter, G. Alber and R. Hoffmann, Api88 Is a Novel Antibacterial Designer Peptide To Treat Systemic Infections with Multidrug-Resistant Gram-negative Pathogens, ACS Chem. Biol., 2012, 7, 1281–1291 CrossRef CAS PubMed.
- R. Schmidt, E. Ostorházi, E. Wende, D. Knappe and R. Hoffmann, Pharmacokinetics and in vivo efficacy of optimized oncocin derivatives, J. Antimicrob. Chemother., 2016, 71, 1003–1011 CrossRef CAS PubMed.
- L. D. Sabath, Six factors that increase the activity of antibiotics in vivo, Infection, 1978, 6, S67–S71 CrossRef.
- N. H. Fedrigo, J. Mazucheli, J. Albiero, D. R. Shinohara, F. G. Lodi, A. C. dos, S. Machado, S. K. B. Sy and M. C. B. Tognim, Pharmacodynamic Evaluation of Fosfomycin against Escherichia coli and Klebsiella spp. from Urinary Tract Infections and the Influence of pH on Fosfomycin Activities, Antimicrob. Agents Chemother., 2017, 61, e02498, DOI:10.1128/AAC.02498-16.
- S. Uttenweiler-Joseph, M. Moniatte, M. Lagueux, A. Van Dorsselaer, J. A. Hoffmann and P. Bulet, Differential display of peptides induced during the immune response of Drosophila: a matrix-assisted laser desorption ionization time-of-flight mass spectrometry study, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11342–11347 CrossRef CAS PubMed.
- P. Wolff, I. Amal, V. Oliéric, O. Chaloin, G. Gygli, E. Ennifar, B. Lorber, G. Guichard, J. Wagner, A. Dejaegere and D. Y. Burnouf, Differential Modes of Peptide Binding onto Replicative Sliding Clamps from Various Bacterial Origins, J. Med. Chem., 2014, 57, 7565–7576 CrossRef CAS PubMed.
- Z. Yin, L. R. Whittell, Y. Wang, S. Jergic, M. Liu, E. J. Harry, N. E. Dixon, J. L. Beck, M. J. Kelso and A. J. Oakley, Discovery of lead compounds targeting the bacterial sliding clamp using a fragment-based approach, J. Med. Chem., 2014, 57, 2799–2806 CrossRef CAS PubMed.
- A. Kling, P. Lukat, D. V. Almeida, A. Bauer, E. Fontaine, S. Sordello, N. Zaburannyi, J. Herrmann, S. C. Wenzel, C. König, N. C. Ammerman, M. B. Barrio, K. Borchers, F. Bordon-Pallier, M. Brönstrup, G. Courtemanche, M. Gerlitz, M. Geslin, P. Hammann, D. W. Heinz, H. Hoffmann, S. Klieber, M. Kohlmann, M. Kurz, C. Lair, H. Matter, E. Nuermberger, S. Tyagi, L. Fraisse, J. H. Grosset, S. Lagrange and R. Müller, Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins, Science, 2015, 348, 1106–1112 CrossRef CAS PubMed.
- P. A. Smith and F. E. Romesberg, Combating bacteria and drug resistance by inhibiting mechanisms of persistence and adaptation, Nat. Chem. Biol., 2007, 3, 549–556 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: SI.1a: HPLC and LCMS analyses of the different peptides synthesized. SI.1b: HRMS analyses of the different peptides synthesized. SI.3: Typical ITC titration curves describing the interaction of the different peptides with EcSC and Ec70S. SI.4: Analysis of the interaction of the different peptides with EcSC and Ec70S. SI.5: Whole gel images of the co-immunoprecipitation assays. SI.6: Survival curves of E. coli infected D. melanogaster from three independent experiments. See DOI: 10.1039/d0cb00060d |
| ‡ These authors contributed equally to this work. |
| § Current address: Polypeptide Laboratories France SAS, 7 rue de Boulogne, 67100 Strasbourg, France. |
| ¶ Current address: Department of Microbiology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, 30-387 Krakow, Poland. |
| This journal is © The Royal Society of Chemistry 2020 |