
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermostabilization of viruses via complex coacervation†
Xue
Mi‡
 ab,
Whitney C.
Blocher McTigue‡
c,
Pratik U.
Joshi
ab,
Mallory K.
Bunker
a,
Caryn L.
Heldt
*ab and
Sarah L.
Perry
*cd
ab,
Whitney C.
Blocher McTigue‡
c,
Pratik U.
Joshi
ab,
Mallory K.
Bunker
a,
Caryn L.
Heldt
*ab and
Sarah L.
Perry
*cd
aDepartment of Chemical Engineering, Michigan Technological University, USA. E-mail: heldt@mtu.edu
bHealth Research Institute, Michigan Technological University, USA
cDepartment of Chemical Engineering, University of Massachusetts Amherst, USA. E-mail: perrys@engin.umass.edu
dInstitute for Applied Life Sciences, University of Massachusetts Amherst, USA
First published on 13th October 2020
Abstract
Widespread vaccine coverage for viral diseases could save the lives of millions of people each year. For viral vaccines to be effective, they must be transported and stored in a narrow temperature range of 2–8 °C. If temperatures are not maintained, the vaccine may lose its potency and would no longer be effective in fighting disease; this is called the cold storage problem. Finding a way to thermally stabilize a virus and end the need to transport and store vaccines at refrigeration temperatures will increase access to life-saving vaccines. We explore the use of polymer-rich complex coacervates to stabilize viruses. We have developed a method of encapsulating virus particles in liquid complex coacervates that relies on the electrostatic interaction of viruses with polypeptides. In particular, we tested the incorporation of two model viruses; a non-enveloped porcine parvovirus (PPV) and an enveloped bovine viral diarrhea virus (BVDV) into coacervates formed from poly(lysine) and poly(glutamate). We identified optimal conditions (i.e., the relative amount of the two polypeptides) for virus encapsulation, and trends in this composition matched differences in the isoelectric point of the two viruses. Furthermore, we were able to achieve a ∼103–104-fold concentration of virus into the coacervate phase, such that the level of virus remaining in the bulk solution approached our limit of detection. Lastly, we demonstrated a significant enhancement of the stability of non-enveloped PPV during an accelerated aging study at 60 °C over the course of a week. Our results suggest the potential for using coacervation to aid in the purification and formulation of both enveloped and non-enveloped viruses, and that coacervate-based formulations could help limit the need for cold storage throughout the transportation and storage of vaccines based on non-enveloped viruses.
Introduction
According to the World Health Organization (WHO), millions of people die from viral infectious diseases each year.1 One of the most effective methods to prevent viral infection is with vaccines. In order for viral vaccines to be effective, they must be transported and stored in a “cold chain”.2 A cold chain is a system of transporting and storing vaccines at the recommended temperature, typically 2–8 °C, from the manufacturer until the point of use.2,3 If temperatures are not maintained, the vaccine may lose its potency and could no longer be effective in fighting disease.4 Approximately half of the vaccines produced each year are discarded due to poor thermal stability.5 The unreliable cold chain system is one of the major causes of inadequate immunization coverage in developing countries.4 Therefore, developing robust, thermostable viral vaccines that are less dependent on the cold chain is urgent and crucial for universal access to immunizations.There are three major types of viral-based vaccines licensed for human use: live attenuated, inactivated, and subunit vaccines.6,7 Live-attenuated vaccines are usually produced by extended passage of a disease-causing (wild) virus in non-human cell culture to weaken the wild virus.8–10 The attenuated virus can still replicate and stimulate high immunity, but has lost the ability to cause disease. Inactivated vaccines are typically treated by chemical or heat inactivation to stop virus replication.8–10 Thus, while inactivated viruses cannot replicate, they can still produce immunogenicity. Subunit vaccines use a component of the virus, such as a surface polysaccharide, capsid protein, or nucleic acid, to stimulate an immune response.8–10 Typically, live attenuated viruses raise the strongest immune response, followed by inactivated viruses, and then subunit vaccines; however, the stability of these three types of vaccines is in reverse order.6,7 Thus, live attenuated viral vaccines tend to be the most sensitive to temperature changes, and tight temperature control is required for them to remain immunogenic.6 There is a need to develop versatile methods to improve the thermal stability of live attenuated vaccines.
Various methods have been developed to create thermostable viral vaccines, ranging from direct genetic modification11,12 to changes in the formulation.13,14 However, genetically modifying a viral vaccine is labour-intensive, virus-specific, and may not be accessible for some targets.11,12 A more standard method to stabilize vaccine formulations is to add stabilizing excipients.13,14 For example, high molar concentrations of sucrose were able to maintain the infectivity and in vivo immunogenicity of an adenovirus serotype 5 at 37 °C for 10 days.13
Further improvements in the thermal stability of virus formulations are often achieved via drying by lyophilization, spray drying, or foam drying.15,16 Drying aims to slow down the physical and chemical degradation of the vaccine. However, these methods typically require the presence of sugars (e.g., sucrose, mannitol, and trehalose), amino acids, and/or other cryoprotectants and bulking agents that tend to hydrogen bond with viral capsid proteins and/or viral envelopes to entrain surface-bound water and form a stabilizing matrix.15–19 For example, lyophilized rotavirus vaccines formulated in optimized buffer conditions with polyvinyl pyrrolidone as a bulking agent, sucrose as a cryoprotectant, and L-arginine and glycine as osmolytes, can retain potency for 20 months at 37 °C and 7 months at 45 °C.18 An analogous strategy where viral encapsulation in hydrated silica was used in place of an organic matrix slowed the infectivity loss of the human enterovirus type 71 by six-fold at 37 °C for 20 days, or at 40 °C for 36 hours.20 Although such formulation methods show promise for thermostabilizing vaccines, the outcomes tend to be the result of large-scale trial and error experiments, and there is a need for a simple, low-cost, and versatile approach for stabilizing viruses.
We propose the use of complex coacervation as a strategy for improving the thermal stability of viral vaccines. Complex coacervation is an associative liquid–liquid phase separation phenomena that results from the electrostatic and entropic interactions between oppositely charged macro-ions.21–24 Complex coacervation has a strong history of use as a method of encapsulation in the food and personal care industries,25–30 and has gained recent attention for use in the fields of drug delivery31–35 and gene therapy.36–38 A number of reports have focused specifically on the incorporation of proteins into complex coacervates, with a goal of protecting proteins against degradation39,40 and potentially enhancing protein thermal stability.41
We recently demonstrated the ability of two-polymer coacervates to effectively encapsulate proteins with a range of different size and charge characteristics.42 Here, we adapt our approach, to study the encapsulation and potential for thermal stabilization of two model viruses (Fig. 1). We characterized the complex coacervation of cationic poly(L-lysine)400 (K400) and negatively charged poly(D,L-glutamic acid)400 (E400) in the presence of a non-enveloped porcine parvovirus (PPV) and an enveloped bovine viral diarrhea virus (BVDV) as a function of the charge ratio of the two polymers present in solution, and quantified the uptake of virus into the coacervate phase. Lastly, we performed accelerated aging studies to characterize the thermal stability of our coacervate-virus formulations as a proof-of-concept for thermostabilizing vaccines of live attenuated viruses.
 | ||
| Fig. 1 Schematic depiction of virus encapsulation via complex coacervation with two oppositely charged polypeptides. | ||
Materials and methods
Materials
Potassium phosphate monobasic (molecular biology grade, ≥99.0%) and sodium chloride (NaCl, ACS grade, ≥99.0%) were a gift from Millipore Sigma (Burlington, MA). Sodium phosphate dibasic heptahydrate (ACS grade, 98.0–102.0%), sodium hydroxide (NaOH, ACS grade, ≥97.0%), sodium dodecyl sulfate (SDS) and dimethyl sulfoxide (DMSO, BioReagent, >99.7%) were purchased from Sigma-Aldrich (St Louis, MO). Hydrochloric acid (HCl, ACS grade, 36.5–38.0%) and (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) (≥99.0%) were purchased from Fisher Scientific (Pittsburgh, PA). Pierce fluorescent dye 5-(and 6)-carboxy-tetramethyl-rhodamine succinimidyl ester (NHS-Rhodamine) was purchased from Thermo Fisher Scientific (Waltham, MA). Thiazolyl blue tetrazolium bromide (MTT) (98%) was purchased from Alfa Aesar (Haverhill, MA). Minimum essential medium (MEM) and Dulbecco's modified Eagle's medium (DMEM) were purchased from Life Technologies (Carlsbad, CA). Polypeptides with a degree of polymerization of 400, poly(D,L-glutamic acid) (E400) and poly(L-lysine) (K400), were purchased from Alamanda Polymers (Huntsville, AL). The polypeptides were used as received without further purification. Characterization information for the polypeptides is given in ESI Table S1.†All aqueous solutions and buffers were prepared using purified water with a resistivity of ≥18 MΩ cm from a Nanopure filtration system (Thermo Scientific, Waltham, MA) and filtered with a 0.2 μm bottle top filter (VWR, Radnor, PA) or a 0.2 μm syringe filter (VWR) prior to use. Phosphate buffered saline (PBS) (pH 7.20 ± 0.03) was prepared by dissolving 0.21 g potassium phosphate monobasic, 0.73 g sodium phosphate dibasic heptahydrate, and 9.0 g NaCl into 1000 mL Nanopure water. Stock solutions of 10 mM polypeptide solutions were prepared on a charged monomer basis and adjusted with 1 M HCl and 1 M NaOH to the desired pH 8.00 ± 0.03 pH units. Zwitterionic buffer solution of 0.4 M HEPES was also adjusted with 1 M HCl and 1 M NaOH to the desired pH 8.00 ± 0.03 pH units.
Virus production, purification, and titration
Porcine kidney cells (PK-13, CRL-6489) and bovine turbinate cells (BT-1, CRL-1390) were purchased from ATCC. Porcine parvovirus (PPV) strain NADL-2, was a gift from Dr. Ruben Carbonell (North Carolina State University, Raleigh, NC). Bovine viral diarrhea virus (BVDV) strain NADL was purchased from USDA APHIS. PPV was propagated in PK-13 cells, and BVDV was propagated in BT-1 cells, as described previously,43,44 and stored at −80 °C until further use. PPV or BVDV were further purified with a Biotech Cellulose Ester 1000 kDa dialysis tubing (Rancho Dominguez, CA) and a BioRad Econo-Pac 10DG desalting column (Hercules, CA) in PBS buffer, as previously described,45 and stored at 4 °C until further use.To determine the concentration of the infectious virus, an MTT assay was used. Briefly, either 8 × 104 cells per mL PK-13 cells in completed MEM media (to titrate PPV)46 or 2.5 × 105 cells per mL BT-1 cells in completed DMEM media (to titrate BVDV)47 were seeded in a clear, flat-bottom, 96-well plate in a volume of 100 μL per well. After one day of incubation, 25 μL per well of virus sample was added to the corresponding host cells in quadruplicate and serially diluted across the plate. After 6 days post-inoculation, 10 μL per well of 5 mg mL−1 MTT reagent in PBS (pH 7.2) was added to the plate. After 4 hours, 100 μL per well of solubilizing agent, 10% SDS in 0.01 M HCl (pH 2.5), was added to the plate. After 12 to 24 hours, plates were read at 550 nm in a Synergy Mx monochromator-based multimode microplate reader (Winooski, VT). The virus dilution that killed 50% of the cells is stated as the virus titer MTT50.44 A similar procedure was used to quantify the cytotoxicity of the coacervates and the individual peptides (see ESI† for details).
Formation of virus-containing complex coacervates
Virus-containing complex coacervate samples were formed by first pipetting water, and then HEPES buffer into a 1.7 mL microcentrifuge tube, followed by the virus (PPV or BVDV), K400, and E400. The samples were vortexed for 5 seconds after the addition of each polypeptide to ensure fast and complete mixing. The recipes for each sample of PPV- and BVDV-containing complex coacervates are detailed in ESI Tables S2 and S3,† respectively. A typical experiment contained a total volume of 240 μL and maintained a constant total polymer concentration of 7 mM (on a monomer basis) while varying the ratio of K400 to E400. The concentration of virus was also maintained constant at 4, 5, and 6 log (MTT50 per mL) for PPV and 4 and 5 log (MTT50 per mL) for BVDV, and all experiments were performed in 10 mM HEPES buffer, pH 8.0. All virus-containing complex coacervates were prepared immediately before use and studied at room temperature. All experiments were performed in triplicate.Virus complex coacervates characterization and quantification
We used turbidity to qualitatively measure the formation of the virus-containing complex coacervates. Briefly, turbidity was measured by placing 100 μL of the sample into a clear, flat-bottom, 96-well plate and measuring the absorbance at 562 nm using a Synergy Mx monochromator-based multimode microplate reader (Winooski, VT).42,48 The measured signal was referenced against a control well containing only Nanopure water and HEPES buffer. Samples were then examined using an Olympus IX51 microscope with a DP72 camera (Center Valley, PA) to confirm the presence or absence of coacervation, and the 100 μL aliquot was recovered for subsequent use in the infectivity assay. Only two concentrations of BVDV coacervates were studied due to the initial concentration of enveloped BVDV propagated being lower than PPV.Viruses were also labelled with a fluorescent dye NHS-Rhodamine that absorbs visible green light at a wavelength of 552 nm and emits orange-red visible light at 575 nm to confirm the presence of the virus in the coacervate phase. 1 mL of purified virus solutions (8 log PPV or 7 log BVDV) were incubated with 10 mg mL−1 NHS-Rhodamine in DMSO solution (2.15 μL for PPV and 6.5 μL for BVDV) for 1 hour at room temperature. Excessive non-tagged fluorescent dye was removed with a BioRad Econo-Pac 10DG desalting column. The fluorescently labelled virus was used immediately to form the virus coacervate, as described above. An aliquot of 100 μL of tagged virus coacervates was transferred to one well of a 96-well plate and examined with an Olympus IX51 microscope. The coacervates droplets were imaged using both brightfield and fluorescence modes and analysed with ImageJ.
An MTT virus infectivity assay was used to quantify the amount of virus present in both the coacervate and the supernatant phases.44 The 240 μL sample containing the complex coacervate and virus in the microcentrifuge tube was centrifuged using an ST16R Centrifuge (Thermo Scientific, Asheville, NC) at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (21475g) for 20 min at 15 °C to separate the supernatant from the dense coacervate phase. Following centrifugation, the supernatant volume was carefully measured and transferred into a new microcentrifuge tube via pipetting. A volume of 220 μL of 2 M NaCl solution was added to the dense coacervate phase (transparent gel) to dismantle the coalesced virus coacervate, followed by vortexing. The concentration of virus in both the supernatant and dismantled coacervate was then titrated by the MTT assay. The volume of the coacervate phase was neglected, though we estimated that the maximum volume of coacervate formed was approximately ∼1 μL. These values were then used to calculate the partitioning of the virus into the complex coacervate phase. The partition coefficient (K) was calculated as:
000 rpm (21475g) for 20 min at 15 °C to separate the supernatant from the dense coacervate phase. Following centrifugation, the supernatant volume was carefully measured and transferred into a new microcentrifuge tube via pipetting. A volume of 220 μL of 2 M NaCl solution was added to the dense coacervate phase (transparent gel) to dismantle the coalesced virus coacervate, followed by vortexing. The concentration of virus in both the supernatant and dismantled coacervate was then titrated by the MTT assay. The volume of the coacervate phase was neglected, though we estimated that the maximum volume of coacervate formed was approximately ∼1 μL. These values were then used to calculate the partitioning of the virus into the complex coacervate phase. The partition coefficient (K) was calculated as:
 | (1) |
Virus thermal stability study
Thermal stability studies were performed using samples where maximal virus partitioning was observed, (i.e., a charge fraction of 0.5 for PPV and 0.6 for BVDV). A microcentrifuge tube containing either the PPV dense coacervate phase or purified PPV was capped and wrapped in Parafilm and put in a digital dry bath (USA Scientific, Ocala, FL) at 60 °C. The BVDV complex coacervate and purified BVDV were similarly put in the dry bath at 40 °C. At each time point, a tube containing ∼1 μL of the dense, virus-containing coacervate sample was removed from the heating block and dismantled in 220 μL of 2 M NaCl. A purified virus sample was also removed from the heat at the same time. The experiment was performed in triplicate. Both samples were then titrated with the MTT assay to determine the remaining infectious virus concentrations. A log reduction value (LRV) of the virus was calculated as: | (2) |
The lifetime of infectious PPV particles τ was determined using a simple model for infectivity loss:13
| n(t) = n0e−t/τ | (3) |
Statistical analysis
Statistical analysis was performed using an unpaired, two-tailed Student's t test. An asterisk (*) denotes p < 0.05 between samples.Results and discussion
The goal of this work was to determine if complex coacervation could be used to encapsulate and thermally stabilize viruses. To this end, we studied the encapsulation and stabilization of two model viruses, non-enveloped PPV and enveloped BVDV. This approach allowed us to explore potential differences between enveloped and non-enveloped viruses.Encapsulation of virus
Previous reports on protein encapsulation using coacervates emphasized the importance of electrostatic interactions in driving protein incorporation.42,52–57 Therefore, it is important to consider the charge state of the viruses used in our study. Table 1 outlines some of the physical properties of the chosen viruses. Notably, both viruses have an acidic isoelectric point,43 meaning that the particles will carry a net negative charge at most physiologically-relevant solution conditions, and will become more negatively charged at higher pH conditions.| Virus | Capsid | Family | Nucleic acid | Size (nm) | pI | Related human viruses | Ref. |
|---|---|---|---|---|---|---|---|
| Porcine parvovirus (PPV) | Non-enveloped | Parvoviridae | ssDNA | 18–26 | 4.8–5.1 | B-19 human parvovirus | 43, 49 and 50 |
| Bovine viral diarrhea virus (BVDV) | Enveloped | Flaviviridae | ssRNA | 40–60 | 4.3–4.5 | Hepatitis C | 43, 49 and 51 |
However, in the context of complex coacervation, we must balance the charge state of our viruses with that of the two complexing polypeptides.42 Therefore, because both PPV58 and BVDV are stable at pH 8.0,59 we elected to perform our experiments at a solution pH of 8.0 using 10 mM HEPES as a neutral, zwitterionic buffer. This solution condition maximizes the negative charge of the virus particles while limiting the loss of charge from the poly(lysine). Furthermore, our previous efforts with proteins had indicated that protein encapsulation decreased dramatically at higher ionic strength conditions.42,53,54 Therefore, experiments were performed in the absence of added salt.
In order to identify optimal conditions for viral encapsulation, we performed coacervation experiments at levels of constant virus concentration and constant total polypeptide concentration while varying the relative amounts of the polycation K400 and polyanion E400. Fig. 2 shows the characteristic optical micrographs of the resulting samples, prepared with the fluorescently labelled virus. Colocalization of the fluorescent signal with the droplets confirmed the successful incorporation of both PPV and BVDV into our complex coacervates. It should be noted that our study did not aim to create a coacervate formulation with a specified droplet size, and the coacervate droplets in our samples coalesce over time. Careful consideration of these types of physical properties would be necessary for translation of this method into actual practice, but are beyond the scope of the current work.
 | ||
| Fig. 2 (a and d) Brightfield, (b and e) fluorescence, and (c and f) merged optical micrographs of (a–c) PPV- and (d–f) BVDV-containing coacervate droplets, demonstrating virus encapsulation. | ||
Turbidity measurements, along with visual inspection via optical microscopy, were used to identify the presence or absence of phase separation (Fig. 3). We observed a general increase in the turbidity signal with increasing virus concentration, consistent with an increase in the total volume of coacervate present, although the qualitative nature of turbidity is such that we cannot decouple an increase in the number of coacervate droplets from changes in droplet size.
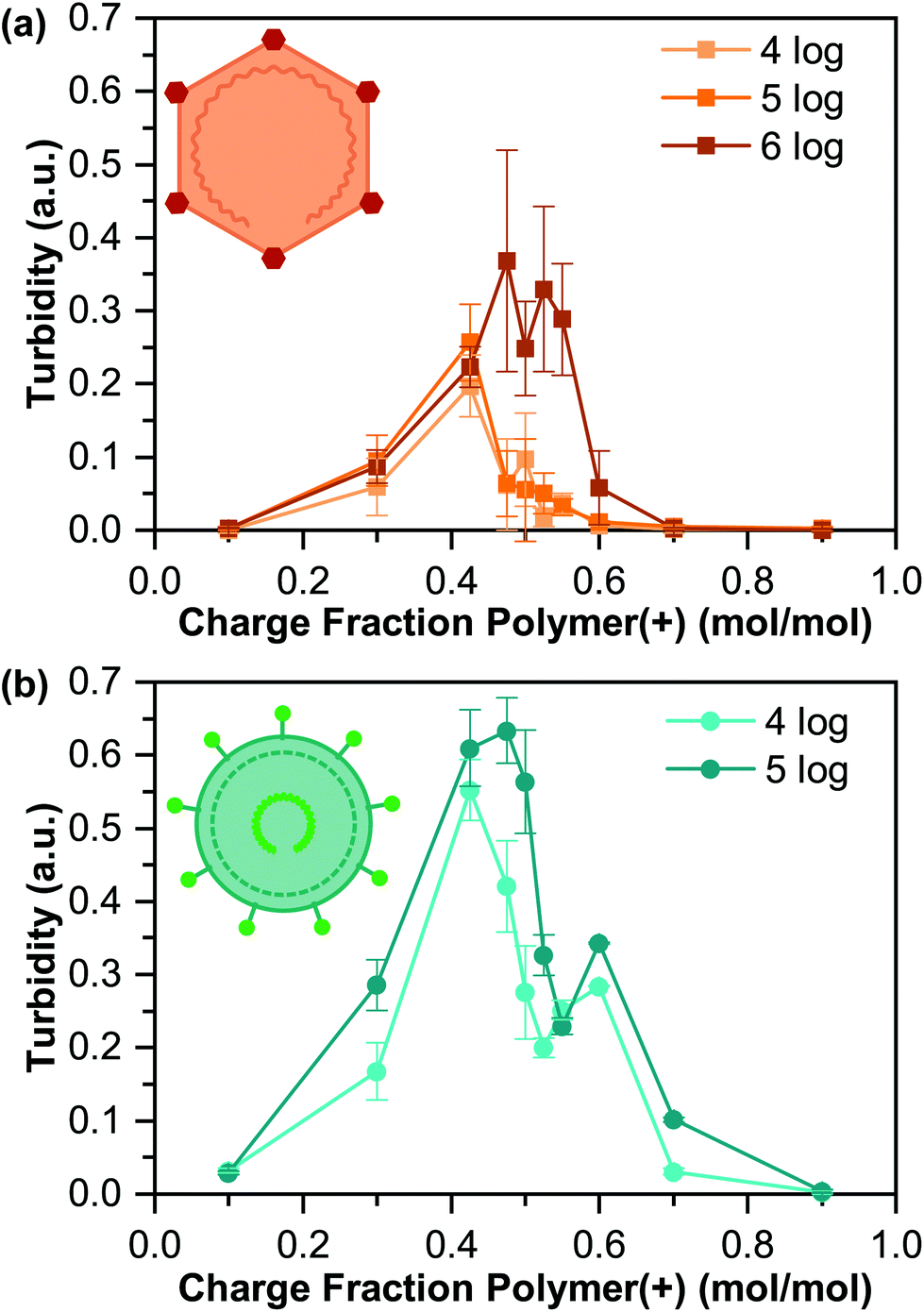 | ||
| Fig. 3 Turbidity of the encapsulated virus as a function of the polymer charge fraction associated with cationic K400 for coacervates prepared with different concentrations of (a) PPV and (b) BVDV. All data points are the average of three separate tests, and error bars represent the standard deviation. | ||
The maximum turbidity signal for all samples was observed at a cationic polymer charge fraction below 0.50, corresponding to “net negative” conditions. This result is somewhat unexpected, as the acidic pI of both viruses would suggest that optimal coacervation would be expected at a “net positive” polymer ratio.42,55,57 However, the turbidity signal for all but one of our samples also showed a bimodal shape, with the second peak located at higher charge fractions. This bimodal signal likely indicates a heterogeneous population of viruses, where each turbidity peak represents a distinct virus population, which is common,60 and could explain the unexpected results.
While optical microscopy and turbidity confirmed the successful formation of virus-containing coacervates, it did not provide quantitative information on the amount of virus sequestered in the coacervates. Therefore, we employed an MTT cell viability assay to quantify the concentration of infectious virus in both the supernatant and coacervate phases. The plots of virus titer as a function of coacervate charge stoichiometry in Fig. 4 showed strong extraction of both PPV and BVDV into the coacervate phase that was matched by a commensurate decrease in the virus titer for the supernatant phase.
 | ||
| Fig. 4 Live virus titration as a function of the polymer charge fraction associated with cationic K400 for both the supernatant (open symbols) and coacervate (closed symbols) phases for samples prepared with different total starting concentrations of (a–c) PPV and (d and e) BVDV. All data points are the average of three separate tests, and error bars represent the standard deviation. | ||
Interestingly, we only observed a single peak in our virus titer data, corresponding to the second peak in the turbidity measurements (i.e., the peak at higher, “net positive” charge fractions). For PPV, this maximum sequestration was observed near a charge ratio 0.5 (Fig. 4a–c), while for BVDV, the maximum occurred near a charge ratio of 0.6 (Fig. 4d and e). This difference in the peak location is likely explained by the fact that pI of BVDV is more acidic than PPV (Table 1), suggesting the potential for a higher net charge at our experimental conditions of pH = 8.0.
Fig. 5 plots the logarithmic value of the partition coefficient ln(K), where a positive value indicates that the viral particles favoured the coacervate phase, while a negative value indicates that the viral particles remained in the supernatant phase. While the trends in partition coefficient derive from those already described in terms of viral titer, what is particularly noteworthy is the magnitude of the partition coefficient. We observed a trend of increasing maximum partition coefficient with the initial viral concentration that achieved 6.2 × 104 fold increase for PPV and 2.4 × 103 fold increase for BVDV. This ability to both sequester and concentrate virus has tremendous potential for applications related to virus purification and formulation. For example, an aqueous two-phase system of poly(ethylene glycol) and salt was only able to achieve an approximate 10-fold partitioning of bacteriophage M13.61
 | ||
| Fig. 5 Partition coefficient as a function of the polymer charge fraction associated with cationic K400 for coacervates prepared with different concentrations of (a–c) PPV and (d and e) BVDV. All data points are the average of three separate tests, and error bars represent the standard deviation. | ||
These observed trends in virus titer and partitioning are also matched by a calculation of the virus recovery into the coacervate phase (ESI Fig. S1†). While our results indicate 100% recovery of PPV into the coacervate phase, we were only able to recover approximately 50% of BVDV. This is likely due to the lower stability of the enveloped BVDV in the high ionic strength conditions used to dissolve the coacervate, as high concentrations of salt can cause leakage in the viral envelope membrane.62 Alternative strategies for destabilizing the coacervate could potentially circumvent this challenge, but are beyond the scope of the current work.
Thermal stability of encapsulated vs. free virus
To demonstrate the effect of complex coacervates on the stability of viruses against high temperatures, we sought to identify accelerated aging conditions for a stability study, using conditions where the viruses would become completely inactivated over a reasonable experimental lifetime. Literature reports on the stability of purified solutions of virus suggested the use of 60 °C for PPV, as a 1 log loss of infectivity was observed for this non-enveloped virus after 1 hour.63 For BVDV, we selected temperature of 40 °C, having observed a 50% loss of infectivity at 37 °C for 6 hours.64We performed stability studies, comparing solutions of free virus in aqueous solution with an equivalent amount of total virus encapsulated in our optimum coacervate conditions. At each time point, a sample of both free and encapsulated virus was removed from heat, the coacervate phase was disassembled by the addition of 2 M NaCl to the coacervate sample, and the viral titer was determined (ESI Fig. S2†). From these data, we calculated the loss of activity over time as a log reduction value (LRV).
For PPV, we observed significant retention of activity due to encapsulation (Fig. 6a). After 1 day at 60 °C, encapsulated PPV effectively maintained its viral titer, only losing 1.0 log ± 0.1 log (MTT50 per mL). In comparison, free PPV showed a LRV of 2.9 log ± 0.3 log (MTT50 per mL) after 1 day. Moreover, free PPV in solution was found to be completely inactivated after 7 days under 60 °C, with an LRV of 5.9 ± 0.5 log (MTT50 per mL), while encapsulated PPV only suffered a titer loss of 2.7 log ± 0.1 log (MTT50 per mL) after 7 days.
 | ||
| Fig. 6 Thermal stability defined as the log reduction value (LRV) as a function of time for free and encapsulated (a) PPV and (b) BVDV. All data points are the average of three separate tests, and error bars represent the standard deviation. Lines are a guide for the eye. An asterisk (*) denotes p < 0.05 comparing encapsulated and free samples at the same time point. | ||
We can further use a simple lifetime model to calculate infectivity loss (eqn (3)). This model assumes the infectious viral particles degraded from infectious to a disrupted state at a constant rate and was fit to the titer data (ESI Fig. S2a†). Based on this model, we determined a lifetime for our encapsulated PPV to be 14 days at 60 °C, which is significantly longer than the 4-day lifetime of free PPV at 60 °C. Using the Simonelli and Dresback's Q10 factor for shelf-life determination and a Q10 value of 2, 7 days at 60 °C is equivalent to 3 months at 22 °C. While a 2 LRV loss would be too much for an FDA approved vaccine, this method shows promise as a method to thermally stabilize non-enveloped viruses at room temperature.65,66
We hypothesize that the enhanced thermal stability of encapsulated PPV compared to the free PPV could be attributed to crowding effects associated with the high concentrations of polymer and virus present in the coacervate. These types of excluded volume effects typically disfavour protein unfolding and denaturation events that could be associated with loss of viral activity.40,67 The main driving force for viral capsid protein unfolding is believed to be the high conformational entropy of the denatured state,68 as the flexible unfolded state has more conformational degrees of freedom than the compact folded state. The limited volume of the crowded coacervate environment would therefore minimize the number of accessible conformational degrees of freedom for the unfolded state, and hence stabilize the native state of the viral protein.68–70 While there is also the potential for enthalpic contributions to protein stability,41,71,72 exploration of these effects would require modulation of the coacervate materials, and is beyond the scope of the current work.
Given the promising improvements in stability seen for PPV (a non-enveloped virus), we similarly explored the stability of BVDV as a model enveloped virus (Fig. 6b and S2b†). However, complex coacervates offered no protection for BVDV against high temperatures. In fact, the data showed that encapsulated samples inactivated faster than free BVDV. We hypothesize that the lipid envelope surrounding the capsid provides a similar entropic stabilization effect for BVDV, as was described in the context of the coacervate for PPV. However, interactions between the coacervate and the lipid bilayer could adversely affect the stability of both the membrane and the virus. Poly(lysine) is known to penetrate negatively charged lipid bilayers,73 and can have cytotoxic effects at high concentrations. However, while a dose-dependent poly(lysine) cytotoxicity was observed for both the PK-13 and BT-1 cells used in this study, it is interesting that the coacervate showed no toxicity with the BT-1 cells used alongside BVDV, while some toxicity was observed for PK-13 cells (ESI Fig. S3†). However, viral envelopes do not play precisely the same role as the membranes of more complex organisms, and potential interference with the BVDV envelope could explain the nearly 2 log difference in initial activity observed for BVDV, as well as the 50% recovery levels of BVDV in coacervates (ESI Fig. S1†). It is possible that these adverse effects could be overcome by a change in coacervate materials and/or experimental methodology. However, such investigations are beyond the scope of the current work.
Conclusions
In summary, we explored the encapsulation of two model viruses, a non-enveloped PPV and an enveloped BVDV, into polypeptide-based complex coacervates. This first proof-of-concept demonstration of viral encapsulation highlighted the tremendous potential for using complex coacervation as a strategy for extracting virus from aqueous solution with near 100% recovery for non-enveloped viruses. Furthermore, strong partitioning of the viruses into the coacervate phase allowed for an increase in virus concentration on the order of 6.2 × 104/2.4 × 103 fold for PPV/BVDV. While these two aspects of downstream viral processing each have significant potential to impact strategies for the purification, concentration, and formulation of viruses, we also demonstrated a significant enhancement in the thermal stability of the non-enveloped PPV. Although more detailed studies on the intermolecular interactions driving these effects is needed, across a range of additional viruses, our results suggest that complex coacervation could help to improve the thermal stability of at least non-enveloped viral vaccines, thereby decreasing the need for a cold chain to maintain their efficacy, decreasing costs, and improving accessibility.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank EMD Millipore for the gift of chemicals used in this work. The authors are grateful to Dr. M. Fernanda Gencoglu for her training on the MTT assay at the University of Massachusetts Amherst and the Institute for Applied Life Sciences Center for Bioactive Delivery for a travel grant. Additional financial support was received from NSF (CAREER-1451959 and CAREER-1945521), the Department of Chemical Engineering at Michigan Tech, and the James and Lorna Mack Chair in Bioengineering.References
- H. Ritchie and M. Roser, Causes of Death, Our World in Data, 2019 Search PubMed, Available from: https://ourworldindata.org/causes-of-death.
- World Health Organization, Immunization in Practice: A practical guide for health staff, World Health Organization, 2015, pp. 1–291 Search PubMed.
- A. Ashok, M. Brison and Y. LeTallec, Improving cold chain systems: Challenges and solutions, Vaccine, 2017, 35(17), 2217–2223, DOI:10.1016/j.vaccine.2016.08.045.
- T. Comes, K. Bergtora Sandvik and B. Van de Walle, Cold chains, interrupted: The use of technology and information for decisions that keep humanitarian vaccines cool, J. Humanit. Logist. Supply Chain Manage., 2018, 8(1), 49–69, DOI:10.1108/JHLSCM-03-2017-0006.
- World Health Organization, Monitoring vaccine wastage at country level: guilines for programme managers, World Health Organization, 2005, pp. 1–77 Search PubMed.
- Centers for Disease Control and Prevention, Epidemiology and Prevention of Vaccine-Preventable Diseases, ed. J. Hamborsky, A. Kroger and S. Wolfe, Public Health Foundation, Washington, D.C., 13th edn, 2015 Search PubMed.
- S. A. Plotkin, Vaccines: the Fourth Century, Clin. Vaccine Immunol., 2009, 16(12), 1709–1719, DOI:10.1128/CVI.00290-09.
- S. J. Flint, L. W. Enquist, V. R. Racaniello, G. F. Rall and A. M. Skalka, Principles of Virology, ASM Press, 4th edn, 2015 Search PubMed.
- HHS and Vaccine Types, 2019, Available from: https://www.vaccines.gov/basics/types.
- NIH and Vaccine Types, 2019, Available from: https://www.niaid.nih.gov/research/vaccine-types.
- C. C. Stobart, C. A. Rostad, Z. Ke, R. S. Dillard, C. M. Hampton, J. D. Strauss, H. Yi, A. L. Hotard, J. Meng and R. J. Pickles, et al., A live RSV vaccine with engineered thermostability is immunogenic in cotton rats despite high attenuation, Nat. Commun., 2016, 7, 1–12, DOI:10.1038/ncomms13916.
- R. Mateo, E. Luna, V. Rincon and M. G. Mateu, Engineering Viable Foot-and-Mouth Disease Viruses with Increased Thermostability as a Step in the Development of Improved Vaccines, J. Virol., 2008, 82(24), 12232–12240, DOI:10.1128/JVI.01553-08.
- M. Pelliccia, P. Andreozzi, J. Paulose, M. D. R. Alicarnasso, V. Cagno, M. Donalisio, A. Civra, R. M. Broeckel, N. Haese and P. J. Silva, et al. Additives for vaccine storage to improve thermal stability of adenoviruses from hours to months, Nat. Commun., 2016, 7(1), 1–7, DOI:10.1038/ncomms13520.
- J. A. White, M. Estrada, E. A. Flood, K. Mahmood, R. Dhere and D. Chen, Development of a stable liquid formulation of live attenuated influenza vaccine, Vaccine, 2016, 34, 3676–3683, DOI:10.1016/j.vaccine.2016.04.074.
- O. S. Kumru, S. B. Joshi, D. E. Smith, C. R. Middaugh, T. Prusik and D. B. Volkin, Vaccine instability in the cold chain: Mechanisms, analysis and formulation strategies, Biologicals, 2014, 42(5), 237–259, DOI:10.1016/j.biologicals.2014.05.007.
- C. J. Burke, T. A. Hsu and D. B. Volkin, Formulation, stability, and delivery of live attenuated vaccines for human use, Crit. Rev. Ther. Drug Carrier Syst., 1999, 16(1), 1–83 CAS.
- S. P. Toniolo, S. Afkhami, A. Mahmood, C. Fradin, B. D. Lichty, M. S. Miller, Z. Xing, E. D. Cranston and M. R. Thompson, Excipient selection for thermally stable enveloped and non-enveloped viral vaccine platforms in dry powders, Int. J. Pharm., 2019, 561, 66–73, DOI:10.1016/j.ijpharm.2019.02.035.
- M. Madan, D. Sikriwal, G. Sharma, N. Shukla, A. K. Mandyal, S. Kale and D. Gill, Rational design of heat stable lyophilized rotavirus vaccine formulations, Hum. Vaccines Immunother., 2018, 1–10, DOI:10.1080/21645515.2018.1487499.
- J. K. Kaushik and R. Bhat, Why Is Trehalose an Exceptional Protein Stabilizer?, J. Biol. Chem., 2003, 278(29), 26458–26465, DOI:10.1074/jbc.M300815200.
- G. Wang, H.-J. Wang, H. Zhou, Q.-G. Nian, Z. Song, Y.-Q. Deng, X. Wang, S.-Y. Zhu, X.-F. Li and C.-F. Qin, et al., Hydrated Silica Exterior Produced by Biomimetic Silicification Confers Viral Vaccine Heat-Resistance, ACS Nano, 2015, 9(1), 799–808, DOI:10.1021/nn5063276.
- W. C. Blocher and S. L. Perry, Complex coacervate-based materials for biomedicine, WIREs Nanomed. Nanobiotechnol., 2017, 9, e1442, DOI:10.1002/wnan.1442.
- Y. P. Timilsena, T. O. Akanbi, N. Khalid, B. Adhikari and C. J. Barrow, Complex coacervation: Principles, mechanisms and applications in microencapsulation, Int. J. Biol. Macromol., 2019, 121(C), 1276–1286, DOI:10.1016/j.ijbiomac.2018.10.144.
- J. Pathak, E. Priyadarshini, K. Rawat and H. B. Bohidar, Complex coacervation in charge complementary biopolymers: Electrostatic versus surface patch binding, Adv. Colloid Interface Sci., 2017, 250(C), 40–53, DOI:10.1016/j.cis.2017.10.006.
- C. E. Sing and S. L. Perry, Recent progress in the science of complex coacervation, Soft Matter, 2020, 50, 9528–9530, 10.1039/D0SM00001A.
- S. L. Turgeon, C. Schmitt and C. Sanchez, Protein–polysaccharide complexes and coacervates, Curr. Opin. Colloid Interface Sci., 2007, 12(4–5), 166–178, DOI:10.1016/j.cocis.2007.07.007.
- C. Schmitt and S. L. Turgeon, Protein/polysaccharide complexes and coacervates in food systems, Adv. Colloid Interface Sci., 2011, 167(1–2), 63–70, DOI:10.1016/j.cis.2010.10.001.
- Y. Yeo, E. Bellas, W. Firestone, R. Langer and D. S. Kohane, Complex Coacervates for Thermally Sensitive Controlled Release of Flavor Compounds, J. Agric. Food Chem., 2005, 53(19), 7518–7525, DOI:10.1021/jf0507947.
- A. Matalanis, O. G. Jones and D. J. McClements, Structured biopolymer-based delivery systems for encapsulation, protection, and release of lipophilic compounds, Food Hydrocolloids, 2011, 25(8), 1865–1880, DOI:10.1016/j.foodhyd.2011.04.014.
- L. Jourdain, M. E. Leser, C. Schmitt, M. Michel and E. Dickinson, Stability of emulsions containing sodium caseinate and dextran sulfate: Relationship to complexation in solution, Food Hydrocolloids, 2008, 22(4), 647–659, DOI:10.1016/j.foodhyd.2007.01.007.
- F. Weinbreck, R. de Vries, P. Schrooyen and C. G. de Kruif, Complex Coacervation of Whey Proteins and Gum Arabic, Biomacromolecules, 2003, 4(2), 293–303, DOI:10.1021/bm025667n.
- B. Sarmento, A. Ribeiro, F. Veiga, P. Sampaio, R. Neufeld and D. Ferreira, Alginate/Chitosan Nanoparticles are Effective for Oral Insulin Delivery, Pharm. Res., 2007, 24(12), 2198–2206, DOI:10.1007/s11095-007-9367-4.
- N. R. Johnson, T. Ambe and Y. Wang, Lysine-based polycation:heparin coacervate for controlled protein delivery, Acta Biomater., 2014, 10(1), 40–46, DOI:10.1016/j.actbio.2013.09.012.
- W. C. Blocher McTigue and S. L. Perry, Protein Encapsulation Using Complex Coacervates: What Nature Has to Teach Us, Small, 2020, 16(27), 1907671–1907617, DOI:10.1002/smll.201907671.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Block Copolymer Micelles in Nanomedicine Applications, Chem. Rev., 2018, 118(14), 6844–6892, DOI:10.1021/acs.chemrev.8b00199.
- H. Kinoh, Y. Miura, T. Chida, X. Liu, K. Mizuno, S. Fukushima, Y. Morodomi, N. Nishiyama, H. Cabral and K. Kataoka, Nanomedicines eradicating cancer stem-like cells in vivo by pH-triggered intracellular cooperative action of loaded drugs, ACS Nano, 2016, 10(6), 5643–5655, DOI:10.1021/acsnano.6b00900.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Polysaccharides-based nanoparticles as drug delivery systems, Adv. Drug Delivery Rev., 2008, 60(15), 1650–1662, DOI:10.1016/j.addr.2008.09.001.
- K. W. Leong, H. Q. Mao, V. L. Truong-Le, K. Roy, S. M. Walsh and J. T. August, DNA-polycation nanospheres as non-viral gene delivery vehicles, J. Controlled Release, 1998, 53(1–3), 183–193, DOI:10.1016/S0168-3659(97)00252-6.
- S. Ozbas-Turan, C. Aral, L. Kabasakal, M. Keyer-Uysal and J. Akbuga, Co-Encapsulation of Two Plasmids in Chitosan Microspheres as a Non-Viral Gene Delivery Vehicle, J. Pharm. Pharm. Sci., 2003, 6(1), 27–32 CAS.
- M. Zhao and N. S. Zacharia, Protein encapsulation via polyelectrolyte complex coacervation: Protection against protein denaturation, J. Chem. Phys., 2018, 149(16), 163326–163311, DOI:10.1063/1.5040346.
- J. J. Water, M. M. Schack, A. Velazquez-Campoy, M. J. Maltesen, M. van de Weert and L. Jorgensen, Complex coacervates of hyaluronic acid and lysozyme: Effect on protein structure and physical stability, Eur. J. Pharm. Biopharm., 2014, 88(2), 325–331, DOI:10.1016/j.ejpb.2014.09.001.
- P. G. Chang, R. Gupta, Y. P. Timilsena and B. Adhikari, Optimisation of the complex coacervation between canola protein isolate and chitosan, J. Food Eng., 2016, 191(C), 58–66, DOI:10.1016/j.jfoodeng.2016.07.008.
- W. C. Blocher McTigue and S. L. Perry, Design Rules for Encapsulating Proteins into Complex Coacervates, Soft Matter, 2019, 15, 3089–3103, 10.1039/C9SM00372J.
- X. Mi, E. K. Bromley, P. U. Joshi, F. Long and C. L. Heldt, Virus Isoelectric Point Determination Using Single-Particle Chemical Force Microscopy, Langmuir, 2019, 36(1), 370–378, DOI:10.1021/acs.langmuir.9b03070.
- C. L. Heldt, R. Hernandez, U. Mudiganti, P. V. Gurgel, D. T. Brown and R. G. Carbonell, A colorimetric assay for viral agents that produce cytopathic effects, J. Virol. Methods, 2006, 135(1), 56–65, DOI:10.1016/j.jviromet.2006.01.022.
- X. Mi, E. M. Lucier, D. G. Turpeinen, E. L. L. Yeo, J. C. Y. Kah and C. L. Heldt, Mannitol-induced gold nanoparticle aggregation for the ligand-free detection of viral particles, Analyst, 2019, 144(18), 5486–5496, 10.1039/C9AN00830F.
- M. F. Tafur, K. S. Vijayaragavan and C. L. Heldt, Reduction of porcine parvovirus infectivity in the presence of protecting osmolytes, Antiviral Res., 2013, 99(1), 27–33, DOI:10.1016/j.antiviral.2013.04.019.
- H. Meng, P. K. Forooshani, P. U. Joshi, J. Osborne, X. Mi, C. Meingast, R. Pinnaratip, J. Kelley, A. Narkar and W. He, et al., Biomimetic recyclable microgels for on-demand generation of hydrogen peroxide and antipathogenic application, Acta Biomater., 2019, 83, 109–118, DOI:10.1016/j.actbio.2018.10.037.
- B. G. Kitchener, J. Wainwright and A. J. Parsons, A review of the principles of turbidity measurement, Prog. Phys. Geogr.: Earth Environ., 2017, 41(5), 620–642, DOI:10.1177/0309133317726540.
- L. C. Norkin, Virology: Molecular Biology and Pathogenesis, ASM Press, 2010 Search PubMed.
- W. S. Weichert, J. S. L. Parker, A. T. M. Wahid, S.-F. Chang, E. Meier and C. R. Parrish, Assaying for Structural Variation in the Parvovirus Capsid and Its Role in Infection, Virology, 1998, 250, 106–117 CrossRef CAS.
- I. S. Kim, Y. W. Choi and S. R. Lee, Optimization and validation of a virus filtration process for efficient removal of viruses from urokinase solution prepared from human urine, J. Microbiol. Biotechnol., 2004, 14(1), 140–147 CAS.
- S. Lindhoud and M. M. A. E. Claessens, Accumulation of small protein molecules in a macroscopic complex coacervate, Soft Matter, 2016, 12(2), 408–413, 10.1039/C5SM02386F.
- S. Lindhoud, L. Voorhaar, R. de Vries, R. Schweins, M. A. Cohen Stuart and W. Norde, Salt-Induced Disintegration of Lysozyme-Containing Polyelectrolyte Complex Micelles, Langmuir, 2009, 25(19), 11425–11430, DOI:10.1021/la901591p.
- S. Lindhoud, R. de Vries, R. Schweins, M. A. Cohen Stuart and W. Norde, Salt-induced release of lipase from polyelectrolyte complex micelles, Soft Matter, 2009, 5(1), 242–250, 10.1039/B811640G.
- A. C. Obermeyer, C. E. Mills, X.-H. Dong, R. J. Flores and B. D. Olsen, Complex coacervation of supercharged proteins with polyelectrolytes, Soft Matter, 2016, 12, 3570–3581, 10.1039/C6SM00002A.
- R. A. Kapelner and A. C. Obermeyer, Ionic polypeptide tags for protein phase separation, Chem. Sci., 2019, 10(9), 2700–2707, 10.1039/C8SC04253E.
- C. S. Cummings and A. C. Obermeyer, Phase Separation Behavior of Supercharged Proteins and Polyelectrolytes, Biochemistry, 2018, 57(3), 314–323, DOI:10.1021/acs.biochem.7b00990.
- H. D. de Arce, L. J. Pérez, S. Castell, M. I. Percedo, P. Domínguez and M. T. Frías, Porcine Parvovirus Infections In Cuba, Rev. Salud Anim., 2009, 31(3), 176–179 Search PubMed.
- K. A. Nascimento, M. L. Mechler, I. R. H. Gatto, H. M. S. Almeida, A. S. Pollo, F. J. F. Sant'Ana, P. M. O. Pedroso and L. G. de Oliveira, Evidence of bovine viral diarrhea virus transmission by back pond water in experimentally infected piglets, Pesq. Vet. Bras., 2018, 38, 1896–1901, DOI:10.1590/1678-5150-pvb-5629.
- E. Valdano, S. Manrubia, S. Gómez and A. Arenas, Endemicity and prevalence of multipartite viruses under heterogeneous between-host transmission, PLoS Comput. Biol., 2019, 15(3), e1006876, DOI:10.1371/journal.pcbi.1006876.
- E. Jue, C. D. Yamanishi, R. Y. T. Chiu, B. M. Wu and D. T. Kamei, Using an Aqueous Two-Phase Polymer-Salt System to Rapidly Concentrate Viruses for Improving the Detection Limit of the Lateral-Flow Immunoassay, Biotechnol. Bioeng., 2014, 111(12), 2499–2507, DOI:10.1002/bit.25316/abstract.
- H.-J. Choi, J.-M. Song, B. J. Bondy, R. W. Compans, S.-M. Kang and M. R. Prausnitz, Effect of Osmotic Pressure on the Stability of Whole Inactivated Influenza Vaccine for Coating on Microneedles, PLoS One, 2015, 10(7), e0134431–e0134422, DOI:10.1371/journal.pone.0134431.
- J. E. A. Blumel, I. Schmidt, H. WIllkommen and J. Löwer, Inactivation of parvovirus B19 during pasteurization of human serum albumin, Transfusion, 2002, 42, 1011–1018 CrossRef.
- K. Depner, T. Bauer and B. Liess, Thermal and pH stability of pestiviruses, Rev. Sci. Tech. – Off. Int. Epizoot., 1992, 11(3), 885–893 CrossRef CAS.
- A. Simonelli, D. Dresback, D. Francke and H. Whitney, Perspectives in Clinical Pharmacy, Drug Intelligence Publications, Hamilton, IL, 1972 Search PubMed.
- S. Y. Lee and J. M. Krochta, Accelerated Sheld Life Testing of Whey-Protein-Coated Peantuts Analyzed bu Static Headspace Gas Chromatography, J. Agric. Food Chem., 2002, 50(7), 2022–2028 CrossRef CAS.
- N. Martin, M. Li and S. Mann, Selective uptake and refolding of globular proteins in coacervate microdroplets, Langmuir, 2016, 32(23), 5881–5889, DOI:10.1021/acs.langmuir.6b01271.
- Y. H. Kim and W. E. Stites, Effects of Excluded Volume upon Protein Stability in Covalently Cross-Linked Proteins with Variable Linker Lengths†, Biochemistry, 2008, 47(33), 8804–8814, DOI:10.1021/bi800297j.
- A. C. Miklos, C. Li, N. G. Sharaf and G. J. Pielak, Volume Exclusion and Soft Interaction Effects on Protein Stability under Crowded Conditions, Biochemistry, 2010, 49(33), 6984–6991, DOI:10.1021/bi100727y.
- J. Ådén and P. Wittung-Stafshede, Folding of an Unfolded Protein by Macromolecular Crowding in Vitro, Biochemistry, 2014, 53(14), 2271–2277, DOI:10.1021/bi500222g.
- A. E. Smith, L. Z. Zhou, A. H. Gorensek, M. Senske and G. J. Pielak, In-cell thermodynamics and a new role for protein surfaces, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(7), 1725–1730, DOI:10.1073/pnas.1518620113.
- M. Sarkar, C. Li and G. J. Pielak, Soft interactions and crowding, Biophys. Rev., 2013, 5(2), 187–194, DOI:10.1007/s12551-013-0104-4.
- Y. Takechi, H. Tanaka, H. Kitayama, H. Yoshii, M. Tanaka and H. Saito, Comparative study on the interaction of cell-penetrating polycationic polymers with lipid membranes, Chem. Phys. Lipids, 2012, 165(1), 51–58, DOI:10.1016/j.chemphyslip.2011.11.002.
Footnotes |
| † Electronic supplementary information (ESI) available: Physical characterization of polymers, sample recipes, virus recovery data, raw virus titer data from thermal stability experiments, and cytotoxicity data. See DOI: 10.1039/d0bm01433h |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2020 |