
Engineering extracellular vesicles for cancer therapy: recent advances and challenges in clinical translation
Sha
Li†
a,
Jinliang
Xu†
 b,
Jun
Qian
c and
Xihui
Gao
*a
b,
Jun
Qian
c and
Xihui
Gao
*a
aKey Laboratory of Medical Molecular Virology (MOE/NHC/CAMS), School of Basic Medical Sciences, Fudan University, 131 Dong An Road, Shanghai 200032, China. E-mail: gaoxihui@fudan.edu.cn
bDepartment of Pharmacology, School of Basic Medical Sciences & State Key Laboratory of Molecular Engineering of Polymers, Fudan University, Shanghai 200032, P.R. China
cSchool of Pharmacy, Fudan University & Key Laboratory of Smart Drug Delivery (Fudan University), Ministry of Education, Shanghai 201203, P.R. China
First published on 13th October 2020
Abstract
Extracellular vesicles (EVs) are receiving increasing attention in recent years in the field of cancer treatment. EVs contain specific contents closely related to their donor cells, such as miRNAs, proteins and dsDNAs. As endogenous vesicles, EVs naturally have the characteristics of low toxicity and low immunogenicity and can stably pass through the circulatory system to reach the recipient cells, which make them good carriers to deliver therapeutic agents such as nucleic acid sequences and chemotherapeutics. In many preclinical studies and clinical trials, EVs have demonstrated their unlimited advantages in the field of cancer therapy. However, there are still some challenges that restrict their clinical application, such as yield, heterogeneity, safety, and specificity. In this review, we will focus on the latest breakthrough of EVs in the field of cancer treatment and discuss the challenges in the clinical translation of EVs.
 Sha Li | Sha Li received her M.S. of Medicine in Clinical Laboratory Diagnostics from Anhui University of Science and Technology in 2020. Currently, she is a research assistant at the School of Basic Medical Sciences, Fudan University. Her research focuses on extracellular vesicle-based nano-structures for cancer diagnosis and therapy. |
 Jinliang Xu | Jinliang Xu received his B.S. degree in Pharmacy from Fudan University in 2020. Currently, he is a Ph.D. student in the Department of Pharmacology, School of Basic Medical Sciences of Fudan University. His research interests focus on the development of targeted drug delivery systems for cancer therapy. |
 Jun Qian | Dr Jun Qian is an associate professor at the School of Pharmacy, Fudan University. She received her Ph.D. in Pharmaceutics in 2014 from the School of Pharmacy, Fudan University. Her research centers on the biomedical applications of radionuclide tracers and the development of molecular probes for tumor diagnosis. |
 Xihui Gao | Dr Xihui Gao received a Ph.D. degree in Pharmaceutics from Fudan University in 2017. He is currently a professor in the Key Laboratory of Medical Molecular Virology (MOE/NHC/CAMS), School of Basic Medical Sciences at Fudan University. His research focuses on the development of biomimetic systems for cancer diagnosis and therapy. |
1. Introduction
Extracellular vesicles (EVs) are lipidic vesicles that can be released from essentially all eukaryotic cell types.1 According to different generation mechanisms, EVs are mainly divided into three subtypes: exosomes (30–100 nm), microvesicles (50–1000 nm) and apoptotic bodies (1000–5000 nm).2–4 Among them, exosomes are generated from the endocytic system. First, the cell membranes are invaded to form the early endosomes, and then the early endosomes develop into the late endosomes (multivesicular bodies, MVBs). During this process, membranes of late endosomes bud inward to form the intraluminal vesicles (ILVs). Finally, MVBs fuse with the cell membranes, and their contents, which are exosomes, are released outside the cells (Fig. 1A).5,6 It is worth noting that in the biogenesis of ILVs, part of the cytoplasmic components will be wrapped into ILVs. These components are not completely encapsulated randomly, but are sorted through the endosomal sorting complex required for transport (ESCRT)-dependent or ESCRT-independent pathways.7,8 Compared with the complex formation of exosomes, microvesicles and apoptotic bodies are formed through direct outward budding of living cell or dying cell membranes.9 In addition, microvesicles also include oncosomes (100–400 nm) released by tumor cells.10,11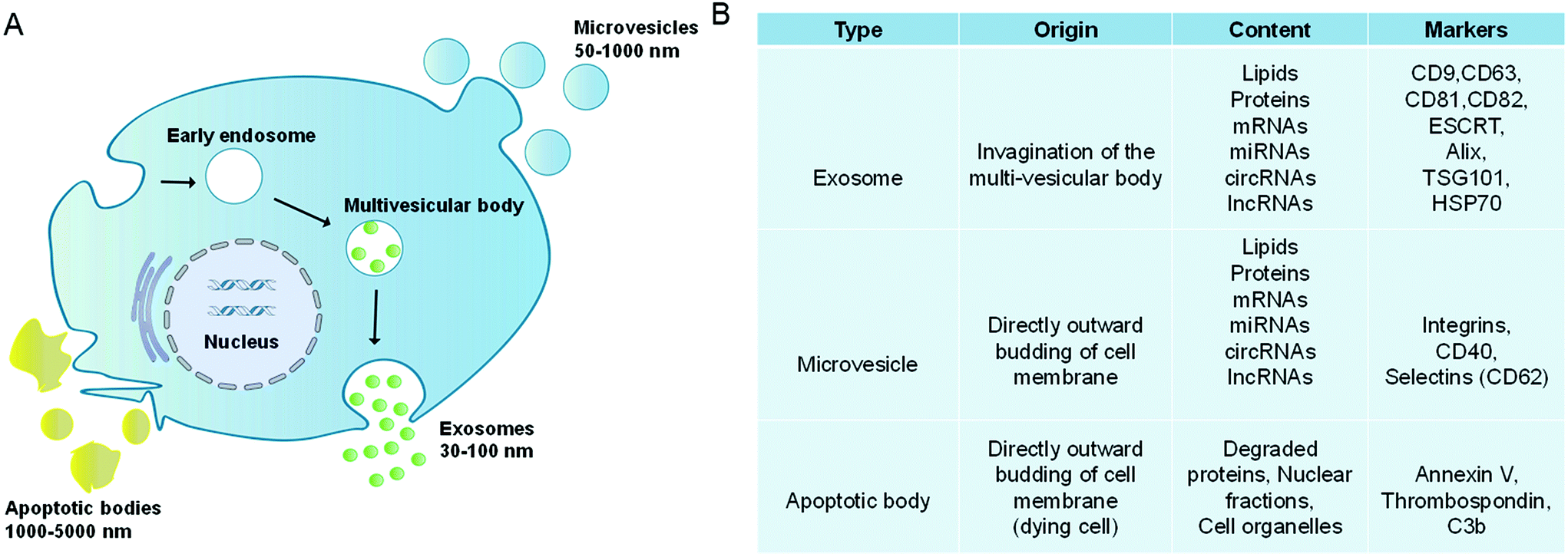 | ||
| Fig. 1 Extracellular vesicles comprise a heterogeneous population of membrane vesicles of various origins. (A) Formation and release mechanisms of extracellular vesicles. (B) Main features of extracellular vesicles. | ||
Due to the different generation mechanisms, the compositions of exosomes and microvesicles are not exactly the same. For example, exosomes are rich in CD9, CD63, CD81, and CD82, as well as ESCRT and HSP70, while microvesicles contain CD40, CD62, etc. (Fig. 1B).12–15 However, both of them contain a lot of the same contents, such as RNAs (mRNAs, lncRNAs, microRNAs and circular RNAs), dsDNAs, integrins, etc.16,17 Besides, they have similar lipid compositions such as sphingomyelin, cholesterol, glycosphingolipids, etc.18–20 In this review, EVs are used as the general term for exosomes and microvesicles. Both apoptotic bodies and oncosomes have their specific cell origins and production mechanisms, which will not be discussed.
EVs not only participate in intercellular communications but also play important roles in various physiological and pathological processes.21 In normal cells, EVs are involved in a series of physiological processes, such as blood coagulation, stem cell differentiation, tissue regeneration and immune regulation.22–25 In tumor cells, EVs, as an important tool for information transmission, can communicate with nearby or slightly distant cells, establish conducive conditions for tumor growth and metastasis and trigger inflammation, angiogenesis, cellular apoptosis, immunosuppression, drug resistance, etc.26–28
The compositions and functions of EVs give them great potential in delivering therapeutic cargos to the target cells. Compared with synthetic vectors, such as liposomes, micelles, and polymeric nanoparticles, EVs are endogenous vesicles that exhibit high biocompatibility and low immunogenicity. Besides, EVs possess enhanced in vivo stability and long blood circulation time, which make them attractive vehicles to deliver therapeutic agents such as nucleic acid sequences and chemotherapeutics. Moreover, the ability of EVs to interact with certain recipient cells gives EVs a natural high targeting efficiency, enabling targeted delivery of therapeutics to specific tissues or tumors. Despite several advantages, EVs also suffer from some drawbacks. For example, unlike synthetic vectors which can be produced in large-scale quantities, the biogenesis of EVs is a natural process. Scaling up the quantities of EVs for therapeutic application poses a major challenge. What's more, EVs are heterogeneous vesicles. The molecular compositions and biological functions of EVs are not only cell-type dependent but also can differ even when the exosomes originate from the same parental cells, which may affect the drug delivery efficiency. In this review, we will focus on the latest breakthrough and clinical applications of EVs in the field of cancer treatment. Meanwhile, the challenges in wider application of the engineered EVs will also be discussed.
2. Isolation and characterization of EVs
EVs exist in a variety of biological fluids including blood, urine, semen, tears and so on. The complex components of these body fluids, such as proteins and nucleic acids, will affect the analysis of EVs. Therefore, it is necessary to isolate EVs from the organism. To date, a variety of isolation methods have been developed, such as ultracentrifugation, density gradient centrifugation, immunoaffinity capture, size-exclusion chromatography, microfluidics, etc.29,30 Ultracentrifugation (UC) is the most widely used method and considered as the gold standard for EV isolation.31 This method can isolate relatively pure EVs, but the high-speed centrifugation will cause the aggregation of EVs and the destruction of EV structures. Thus, the ultrafiltration and the density gradient centrifugation methods have been derived and developed.32,33 Both the two methods can further improve the purity of EVs and avoid protein contamination, but there are still the problems of long extraction times and low yields. Recently, Duong et al. developed cushioned-density gradient ultracentrifugation (C-DGUC) based on the principle that different membrane vesicles have different floatation speeds and equilibrium densities.34 This method improved the recovery rate of EVs by reducing the aggregation of EVs and unstable EV pellets. The immunoaffinity capture method can capture EVs via antibody-modified microbeads or plates. The commonly used markers are CD63 and CD81. This method can isolate various subgroups of EVs, but issues such as low separation efficiency and high costs come up. Microfluidic platforms are devices that can be used to isolate EVs. They possess the advantages of fast processing, cost-effectiveness and accurate classification, but only a small amount of specimens can be isolated each time.35Asymmetric-flow field-flow fractionation (AF4) is a new representative method for EV separation.36 By optimizing the conditions of AF4, subgroups of exosomes can be successfully separated from the total EVs. This technology was widely utilized in the pharmaceutical industry, such as the detection of biological macromolecules and the characterization of nanoparticles. Besides, combination of several methods may achieve a better effect in the separation of EVs. Benedikter et al. developed an isolation strategy (UF-SEC) by orderly combining ultrafiltration and size-exclusion chromatography.32 The results demonstrated that UF-SEC could separate EVs with sufficient purity and high concentration, which meets the requirements of the next compositional and functional characterization. UF-SEC reduced the conglomeration of EVs, which is a main disadvantage of UC. In summary, multifarious methods and technologies have been developed to isolate and purify EVs, thereby promoting the subsequent usage of EVs. The emergence of every new technology or approach may lead to a breakthrough in this area. However, each of them has its drawbacks, leading to expanded difficulties in obtaining high-purity EVs with high efficiency.
Generally, the characterization of EVs often focuses on measuring the particle size and number, as well as detecting specific surface proteins and cargos.37 Transmission electron microscope (TEM) imaging is a standard method to characterize the structure and size of EVs. Besides, scanning electron microscopy (SEM) and dynamic light scattering (DLS) are also used to characterize EVs. Nanoparticle tracking analysis (NTA) can calculate the size and concentration of EVs through tracking and analyzing the Brownian motion of particles.38 NTA is commonly used in counting EVs, but this method cannot distinguish EVs and non-EVs.39 Flow cytometry (FCM) is also a popular way for EV analysis. Imaging flow cytometry (IFCM) is a more effective method derived from traditional FCM. It combines FCM and imaging functions to collect signals and quantify them via the images detected with charge-coupled device (CCD) cameras. Görgens et al. reported that the IFCM could simultaneously detect single EVs and cells in unprocessed samples.40 Compared to normal FCM, IFCM exhibited a lower signal-to-noise ratio and higher overall sensitivity. Fluorescence-activated cytometric sorting (FACS) can identify the surface proteins of EVs, but its resolution is generally above 500 nm.41 Therefore, EVs with a particle size of less than 500 nm must be adsorbed to large-sized microspheres before detection, which complicates the procedure.42
3. Engineered EVs for cancer therapy
EVs released from donor cells can target recipient cells for information transmission. Moreover, EVs also can cross the physiological barriers, such as the blood–brain barrier. These characteristics enable EVs as promising candidates in tumor treatment. Natural EVs shed by cell types can be directly used as tumor therapeutic agents. For example, EVs derived from dendritic cells are rich in histocompatibility complex class I and class II/peptide complexes, which can trigger other immune system cell types and activate antitumor immune responses.4 Similarly, EVs released from natural killer cells mediate the process of anti-melanoma.43 However, when EVs obtained through these methods are directly used as tumor therapeutic agents, they still face the challenges of unclear production mechanisms, difficult quantitation of obtained products, uncontrollable production processes, low yield, and the mutable contents of EVs due to the change of parental cells. In contrast, based on the inherent properties of EVs, engineering EVs as a drug delivery system (DDS) is a more promising strategy for tumor treatment. There are two main strategies to encapsulate therapeutic cargos into EVs. One strategy is to engineer parental cells to shed EVs containing specific therapeutic cargos, whereas the other strategy is to engineer EVs directly via loading therapeutic cargos.Under the stimulation of biological, chemical, or physical methods, the parental cells could yield EVs containing therapeutic agents. For example, Usman et al. developed a method to deliver therapeutic RNA (including antisense oligonucleotides, Cas9 mRNA, and guide RNAs) by mass production of RBC derived EVs (RBCEVs) (Fig. 2A). Subsequently, they validated this delivery strategy on leukemia cells and breast cancer cells. The RNA drugs delivered by RBCEVs showed highly effective miRNA inhibition and CRISPR-Cas9 genome editing with no cytotoxicity.44 Transfection of donor cells with overexpressed genes and then packaging the expression products into EVs is another method to encapsulate therapeutic agents in EVs via engineering parental cells. This method can continuously produce EVs containing therapeutic cargos, whereas the EV membranes can remain intact. For example, O'brien et al. developed engineered mesenchymal stem cells (MSCs) overexpressing miR-379, and then obtained miR-379-rich EVs, which could inhibit the development of metastatic breast cancer.45 HEK-293 T cells transfected with CD-UPRT fusion cassette could secrete EVs rich in suicide molecules CD-UPRT. After these EVs entered glioblastoma cells, the CD-UPRT fusion protein released by them converted the prodrug 5-FC into 5-FU, which caused cell death and significantly inhibited the growth of glioblastoma (Fig. 2B).46 Small-molecule nanoparticles and proteins can also enter cells by direct incubation with cells or electroporation to be further encapsulated into EVs. Generally, in this method, the parental cells are treated with high exposure of cargos, whereas only a small part of them can enter the EVs eventually. However, Cheng et al. developed and purified synthetic multivalent antibodies retargeted exosomes (SMART-Exos) via transfection of plasmids. The SMART-Exos express both monoclonal antibodies, which are specific for T-cell CD3 and cancer cell-associated epidermal growth factor receptor (EGFR). As shown by the results, these exosomes specifically bound to triple-negative breast cancer cells expressing both T cells and EGFR, resulting in an enhanced antitumor effect both in vitro and in vivo.47 Similarly, Yuan et al. developed engineered MSCs via transduction of lentiviruses expressing human TRAIL. The TRAIL-loaded EVs isolated from the engineered MSCs induced apoptosis in 11 cancer cell lines in a dose-dependent manner.48
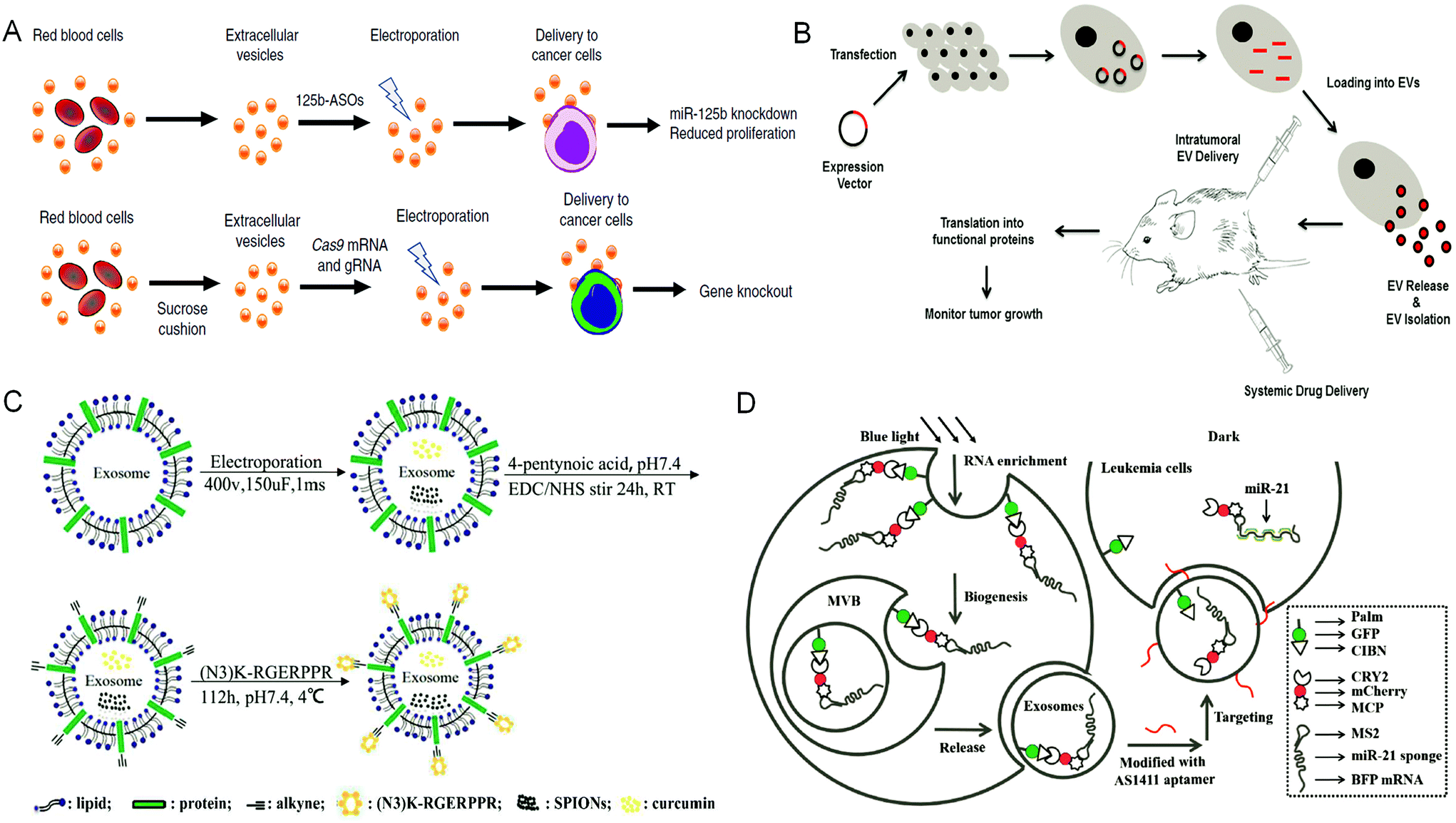 | ||
| Fig. 2 Engineering extracellular vesicles for cancer therapy. (A) Efficient RNA delivery using red blood cell extracellular vesicles. Reprinted with permission from ref. 44, Copyright 2018, Springer Nature. (B) Genetically engineered parental cells to produce extracellular vesicles with specific expression contents, Ref. 46. (C) The preparation of RGE-Exo-SPION/Cur through a click chemistry reaction. Reprinted with permission from ref. 52, Copyright 2018, Elsevier. (D) Construction of a recombinant exosome system that can deliver miR-21 sponges to target tumor leukemia. Reprinted with permission from ref. 56, Copyright 2019, John Wiley and Sons, Inc. | ||
Specifically, Yang et al. developed a cellular-nanoporation biochip to produce exosomes containing therapeutic nucleotide sequences. Compared with other methods such as electroporation, this strategy achieved higher secretion of exosomes and enhanced transcript of RNAs of interest, greatly simplifying the entire process of therapeutic exosome production. Besides, they also demonstrated that the exosomes containing mRNA produced by this strategy restored the tumor-suppression function in orthotopic glioma mouse models.49
Direct engineering of EVs is another efficient way to load therapeutic cargos. Hydrophobic cargos, such as DOX, could be loaded to EVs by passive incubation. For hydrophilic cargos, it is necessary to load them through physical or chemical methods such as sonication, freeze–thaw cycles, saponin treatment, extrusion and electroporation. Many engineering methods focused on modifying the surface of EVs to improve the performance of EVs, among which click chemistry is widely used owing to its simple operation and water and oxygen insensitivities. Meanwhile, it does not need chromatographic purification and almost has no influence on the structure and size of EVs.50,51 Jia et al. have prepared RGE-modified, SPION and Cur-loaded exosomes via electroporation and click chemistry for the diagnosis and treatment of glioma (Fig. 2C).52
Molecules can also be inserted directly into the phospholipid bilayer structures of EVs. In a recent work, we developed a DDS based on engineered macrophage-derived exosomes, which were loaded with PLGA/DOX nanoparticles and can be used for targeted chemotherapy of triple-negative breast cancer (TNBC). To improve the targeting ability of this delivery system, polypeptides were inserted on the surface of exosomes. These peptides can target the mesenchymal-epithelial transition factor overexpressed on the surface of TNBC cells. The results showed that these engineered exosomes significantly improved the uptake efficiency and anti-tumor efficacy of DOX.53
Besides, hybridization is another effective method for engineering EVs. Lin et al. incubated the original exosomes with liposomes and consequently encapsulated the CRISPR/Cas9 system into hybrid exosomes. Therefore, the hybrid exosomes could be endocytosed by MSCs and express the encapsulated genes, which is promising in in vivo gene manipulation.54 Similarly, Piffoux et al. developed a method of utilizing polyethylene glycol to trigger the fusion of EVs with functionalized liposomes. The hybrid EVs exhibited an improved encapsulation rate of exogenous lipophilic or hydrophilic compounds, with similar intrinsic content and biological properties as before. Functionally, they could achieve higher efficiency of drug delivery to cancer cells.55
Parental cells and their secreted EVs can also be co-engineered to produce aimed EVs. Huang et al. engineered the exosome producer cells to enrich miR-21 sponges (a kind of miR-21 inhibitor) through a reversible light-inducible strategy. Then they modified exosomes with the cholesterol-conjugated aptamer AS1411 which could target leukemia cells and block the function of MiR-21 (Fig. 2D).56 Liu et al. cultured HEK293 T cells with DOX-containing medium to make cells secrete EVs containing DOX.57 The surface of EVs was further modified with lipidomimetic chain conjugated HA (LipHA), consequently forming lipHA-engineered hEVs (lipHA-hEVs) loaded with DOX. EVs (hEVs) secreted by 293 T cells can reduce the expression of P-glycoprotein (P-gp) in drug-resistant MCF7/ADR cells. The modification of LipHA increased the tumor-targeting ability of hEVs, which significantly promoted the accumulation of DOX in resistant breast cancer cells. Jung et al. developed and purified hypoxia-targeted theranostic exosomes by exposing the parental cells to hypoxia. The exosomes were modified to carry olaparib, a PARP inhibitor by electroporation. As shown by the results, these exosomes exhibited preferential intake by tumor hypoxic cells and lead to increased apoptosis and inhibited tumor growth in vivo.58 Ingato et al. utilized sulfhydryl-blocking to induce the production of nanovesicles by EL4 (murine lymphoma) cells and then encapsulated them with DOX via incubation (Fig. 3A).59 These nanovesicles achieved efficient and safe delivery of drugs to cancer cells. Moreover, Kamerkar et al. firstly transformed the parental cells via transfection. Thus, the membranes of secreted exosomes are enriched with CD47, a protein which could inhibit the endocytosis of exosomes, consequently prolong the circulation time and reduce the clearance of them. Later, they loaded the exosomes with siRNA or shRNA via electroporation, which are specific to oncogenic KRASG12D, a common mutation of pancreatic cancer. These engineered exosomes exhibited a tumor suppression effect in multiple mouse models of pancreatic cancer, with prolonged survival time.60 Similarly, Hong et al. transfected HEK293 T cells with a plasmid encoding full-length PH20 hyaluronidase which can decompose hyaluronan overexpressed in the tumor microenvironment and suppress tumor growth. And the exosomes with PH20 were loaded with DOX via co-incubation, exhibiting a combination anti-tumor effect.61
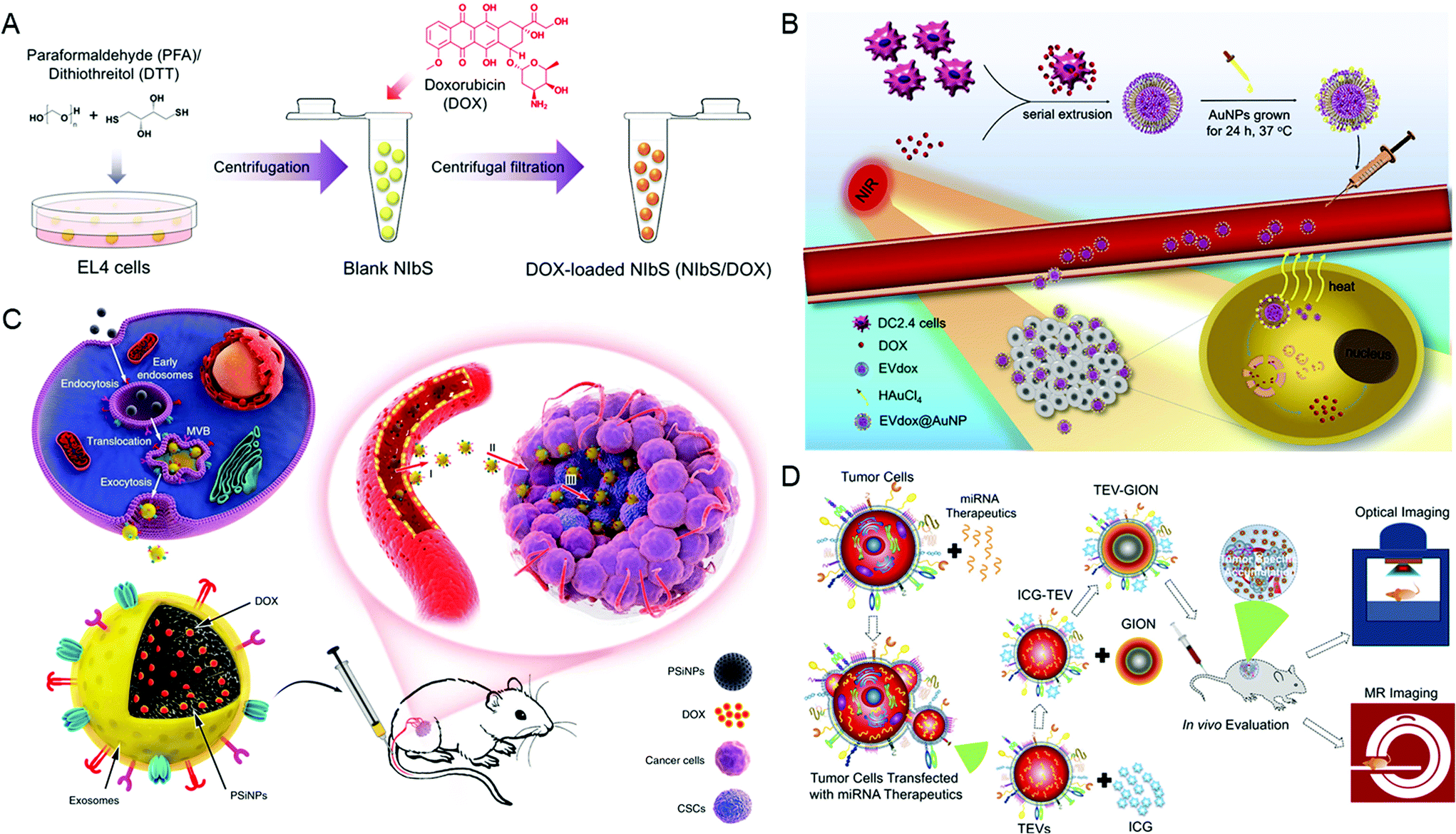 | ||
| Fig. 3 Engineering extracellular vesicles for cancer therapy. (A) Sulfhydryl-blocking induced nanovesicles for efficient drug delivery. Reprinted with permission from ref. 59, Copyright 2018, American Chemical Society. (B) Self-grown gold nanopopcorn for combinatorial chemo-photothermal therapy. Reprinted with permission from ref. 62, Copyright 2019, Elsevier. (C) Tumor-cell-exocytosed exosome-biomimetic porous silicon nanoparticles (PSiNPs) for targeted cancer chemotherapy. Reprinted with permission from ref. 63, Copyright 2019, Springer Nature. (D) Extracellular vesicle-coated nanocarriers for the targeted delivery of anti-miR-21. Reprinted with permission from ref. 64, Copyright 2018, American Chemical Society. | ||
Finally, there are still a variety of research studies which have developed EV-based mimetics for cargo delivery.62 These mimetics not only retained the characteristics of EVs but also received other advantages such as higher tumor-targeting ability (Fig. 3B). Yong et al. developed biocompatible tumor-cell-exocytosed exosome-biomimetic porous silicon nanoparticles (PSiNPs) as drug carriers for targeted cancer chemotherapy (Fig. 3C). As reported, exosome-sheathed PSiNPs could effectively deliver DOX to both bulk cancer cells and cancer stem cells via intravenous administration, resulting in strong anti-tumor efficacy.63 Similarly, Bose et al. have reported tumor cell-derived EV-coated nanocarriers (Fig. 3D). Firstly, they designed and purified EVs containing anti-miR-21, which were shown to attenuate DOX resistance in breast cancer cells. And then, they functionalized the gold–iron oxide nanoparticles with these EVs (TEV-GIONs). The TEV-GIONs were demonstrated to specifically accumulate at tumor sites and exhibited an anti-tumor effect in combination with DOX.64
4. On the way to clinical application
It has been proved that EVs play an important role of bio-modulatory during normal and pathological processes. Meanwhile, the number of circulating EVs and their ingredients will change as the disease progresses. Currently, they have been widely utilized in disease-related fields. Some of them even have moved towards clinical trials (Table 1). Here, some representative studies were discussed.| Indication | Phase and number | Source | Manipulation | Administration | Results/outcome measures | Ref. |
|---|---|---|---|---|---|---|
| DCs: dendritic cells; SC: subcutaneous injection; ID: intradermal injection; DTH: delayed-type hypersensitivity; PFS: progression-free survival; OS: overall survival; IT: intrathoracic injection; GM-CSF: granulocyte-macrophage colony-stimulating factor; QOD: every other day; MTX: methotrexate; PEM: pemetrexed; DDP: cisplatin; TMPs: tumor microparticles; MSCs: mesenchymal stromal cell exosomes; PTBD: percutaneous transhepatic biliary drainage. | ||||||
| Melanoma, stage IIIB and IV metastatic melanoma | Phase 1, n = 15 | Autologous immature DCs | MAGE-3 loaded exosomes | SC and ID | Toxicity: <grade II | 69 |
| Weekly, four weeks | No MAGE3 specific CD4+ and CD8+ T cell responses in peripheral blood | |||||
| Non-small cell lung cancer | Phase 1, n = 13 | Autologous, immature DCs | Exosomes loaded with MAGE-A3, -A4, -A10, and MAGE-3DPO4 peptides | SC and ID | Toxicity: grade 1–2 | 68 |
| Stage III A or B or stage IV, unresectable | Weekly, four weeks | 9 completed therapy, DTH reactivity against MAGE peptides in 3/9, MAGE-specific T cell responses in 1/3, increased NK lytic activity in 2/4 | ||||
| Non-small cell lung cancer | Phase 2, n = 22 | Autologous, IFN-γ matured DCs | Tumor-peptide loaded exosomes | Four ID at 1-week interval | Toxicity: one patient exhibited a grade three hepatotoxicity | 71 |
| Stage III B or stage IV, unresectable | Median PFS 2.2 months and median OS 15 months | |||||
| Colorectal cancer stage III or IV | Phase 1, n = 40 | Autologous, malignant ascites | Unmodified exosomes, ±GM-CSF | Four SC at weekly intervals | Toxicity: grade 1–2 tumor-specific antitumor cytotoxic T lymphocyte (CTL) response in the group treated with exosomes plus GM-CSF (10/13) | 72 |
| Malignant pleural effusion (MPE) | Randomized, parallel controlled trial, n = 62 | A549 | MTX loaded TMPs | IT, QOD, from 5 to 15 days after PEM, DDP (IV) at day 1 in 21-day cycles; control group: saline | Significant improvement of MPE and neutrophil recruitment in the group treated with MTX loaded TMPs. | 74 |
| Obstructive extrahepatic cholangiocarcinoma (ECCA) end-stage | Phase 1, n = 20 | HL-60 | MTX loaded TMPs | Injected into the bile-duct lumen after PTBD | Toxicity: most patients (about 70%) had a transient fever (1–4 h) but no other uncomfortable symptoms | 75 |
| Relief of obstruction in 5/20 | ||||||
| End-stage lung cancer with metastatic MPE multi-drug resistance | Phase 1, n = 6 | A549 | Cisplatin-loaded TMPs | IT for 1–4 weeks | >95% tumor cells in the malignant fluids disappeared, improved symptoms in patients treated with cisplatin-loaded TMPs | 76 |
| Control group (n = 3): IT cisplatin alone | ||||||
| Colon cancer | Phase 1, n = 7 | Plant | Curcumin loaded exosomes | Three arms with curcumin tablets, curcumin with plant exosomes, and no treatment | Primary outcome measures: concentration of curcumin in normal and cancerous tissue | NCT01294072 |
| Metastatic pancreas cancer, with KrasG12D mutation | Phase 1, n = 8 (estimated enrollment) | MSCs | KRASG12D siRNA-loaded exosomes | Received for 15–20 minutes on days 1, 4, and 10. Treatment repeats every 14 days for up to 3 courses | Primary outcome measures: maximum tolerated dose determined by dose limiting toxicity and other 3 additional courses | NCT03608631 |
| Head and neck cancer | Phase 1, n = 60 | Grape | Grape extract | Two arms with grape extract administered orally and standard oral mucositis therapy | Primary outcome measures: pain caused by oral mucositis | NCT01668849 |
| Oral mucositis | ||||||
In some clinical trials, EVs have been developed to deliver different cargos such as tumor antigens, RNAs and drugs, for achieving various applications. An important application of EVs in the clinic is tumor vaccination. For example, an initial study has been conducted on specific vesicles derived from dendritic cells (DCs), termed dexosomes. DCs are antigen-presenting cells (APCs), which process antigens and present them to T-cells by complexing antigens with major histocompatibility complexes (MHCs) and displaying them on the surfaces. However, the results of characterization showed that dexosomes also enwrapped the necessary components of antigen presentation, including the MHC-antigen peptide complexes and immunostimulatory factors.65 Functionally, they played an important role in the stimulation of T-cells via transferring these necessary components from stimulated DCs to antigen-naïve DCs.66 These findings proposed a possible mechanism of DEX in amplifying immune response, which might be translated into the immunotherapy of cancer. The DEX performed as expected in a mouse tumor model. It provoked specific cytotoxic T cells in vivo, inducing growth inhibition and eradication of the established murine tumors.67 In a completed phase I clinical trial, dexosomes loaded with tumor antigens (termed DEX) have been administered to the patients with advanced NSCLC. As shown by the results, 3 of 9 patients achieved tumor-specific systemic immune responses and the DEX slowed the progression of NSCLC in some patients with prolonged stabilization of disease. Assessment of safety demonstrated its great tolerance as no significant toxicity and no detectable autoimmune reaction were observed. Meanwhile, the results of the preliminary test showed that DEX therapy increased the activity of NK cells. As limited by the sample size, these outcomes required a further clinical trial to support.68 Another clinical trial utilized DEX as immunotherapy for metastatic melanoma. In this study, 15 patients with biopsy-proven stage IIIB and IV metastatic melanoma expressing MAGE3 antigen were included. The MAGE3 loaded DEX resulted in objective response of one patient, exhibiting prolonged stability of disease for 24 months. Besides, the investigators detected no severe toxicity as well as no delayed-type hypersensitivity response to DEX, indicating the security of this vaccine. Similar to previously reported trials mentioned before, they observed an enhanced effect of NK cells, proposing the connection between DEX therapy and NK cell activation.69 Since the two phase I trials, a second generation of DEX (IFN-g-Dex) has been established via updated technology, which can lead to enhanced T cell response.70 In a phase II trial, IFN-g-Dex was administered to 22 patients with unresectable NSCLC who have already received 4 cycles of first-line platinum-based chemotherapy. Only one of the 22 patients exhibited a grade three hepatotoxicity, whereas most of them (86%) showed no significant toxicity reaction. The median progression-free survival (PFS) of all patients was 2.2 months after 4 months of therapy. And the median overall survival (OS) reached 15 months with 86% survival rate at 6 months. However, a distinct discovery was that the IFN-g-Dex did not induce a significantly adaptive immune response to tumor antigen, but only increased the function of NK cells. The further study highlighted the relationship between enhanced NK cells and prolonged survival of patients and explained the possible mechanism of this phenomenon.71
Dai et al. reported a phase I trial that focused on the feasibility and safety of autologous ascites-derived exosomes (Aex) combined with granulocyte-macrophage colony-stimulating factor (GM-CSF) for colorectal cancer.72 Previously, another clinical trial utilized GM-CSF as an adjuvant added to peptide vaccine in patients with resected melanoma.73 There is also a previous finding that the Aex could induce effective anti-tumor immunity in a mouse model, which could be increased by the GM-CSF. And this clinical trial further proved that patients treated with Aex or Aex with GM-CSF exhibited great tolerance as no severe toxicity appeared, indicating the acceptable safety of these administrations. Meanwhile, although both Aex and Aex plus GM-CSF could induce antigen-specific anti-tumor immunity, the addition of GM-CSF induced higher production and enhanced toxicity of CTL and promoted systemic anti-tumor immunity. Together, they demonstrated that the Aex could be utilized as a vaccine of CRC, whereas its efficiency could be improved by the GM-CSF adjuvant.
In addition to delivering tumor-specific antigens, several clinical trials proved that the EVs could also serve as a DDS for immunotherapy. Xu et al. developed methotrexate (MTX)-packaging, tumor cell-derived micro-particles (MTX-TMP) for curing malignant pleural effusion (MPE), a common complication of various cancers.74 After perfusion via a pleural catheter, they discovered neutrophil recruitment in the effusions from patients diagnosed with non-squamous NSCLC with primary MPE. Compared with the control group (treated with saline), the experimental group exhibited a significant decrease of MPE. They further demonstrated the positive reaction of neutrophil recruitment in improving MPE. Generally, it provided a possible strategy for mobilizing the innate immune system of patients, especially for activating the neutrophils in the control of malignant fluids. Furthermore, in another clinical trial, Gao et al. gave MTX-TMP to patients with end-stage extrahepatic cholangiocarcinoma (ECCA) (n = 20) with malignant biliary obstruction to study its treatment efficacy.75 After treatment via MTX-TMP perfusion for 7 days (once daily), obstruction of 5 patients was relieved, and one of them even achieved a long remission period of 5 months. All patients did not show severe treatment-related adverse reactions. Further, they demonstrated that the MTX-TMP could recruit neutrophils to the bile, where the neutrophils facilitate the absorption of MTX-TMP by CCA cells. They explained that the anti-tumor effect was obtained by the cooperation of anti-tumor neutrophils activated by environmental MTX-TMP and the pyroptosis induced by tumor-internal MTX-TMP. Together, they provided a novel neutrophil-targeting immunotherapeutic agent with both direct and indirect anti-tumor effects. Moreover, the drug loaded TMPs have been demonstrated to reverse the drug resistance of tumor-repopulating cells.76 This clinical trial recruited six end-stage lung cancer patients with metastatic MPE; the primary tumor cells in their malignant fluids exhibited resistance to cisplatin. Half of them were treated with cisplatin-loaded TMPs via intrathoracic injection, whereas the others were injected with free cisplatin as the control. Following treatment, of course, the control group did not exhibit any improvement owing to the cisplatin-resistance. But in the experimental group, 95 percent of tumor cells in the malignant fluids disappeared, accompanied by the mitigation of the MPE. Besides, the increased survival of the experimental group comes without a significant side effect.
Despite the completed clinical trials, there are still several clinical trials underway. One clinical trial (NCT01294072) investigates the ability of plant exosomes to deliver curcumin to normal and colon cancer tissue. Another phase I trial (NCT03608631) aims to investigate the best dose and safety of KRASG12D siRNA-loaded mesenchymal stromal cell-derived exosomes, which focused on the patients suffering from pancreatic cancer with KrasG12D mutation that has spread to other places in the body. Besides, a distinct study (NCT01668849) focuses on the anti-inflammatory ability of oral grape exosomes to prevent oral mucositis during radiation and chemotherapy treatment for head and neck tumors.
5. Challenges of EV-based cancer therapy
Although some positive outcomes have been achieved in current clinical trials, there are still several barriers that need to be overcome to implement the wide clinical translation of extracellular vesicles in cancer therapy (Fig. 4).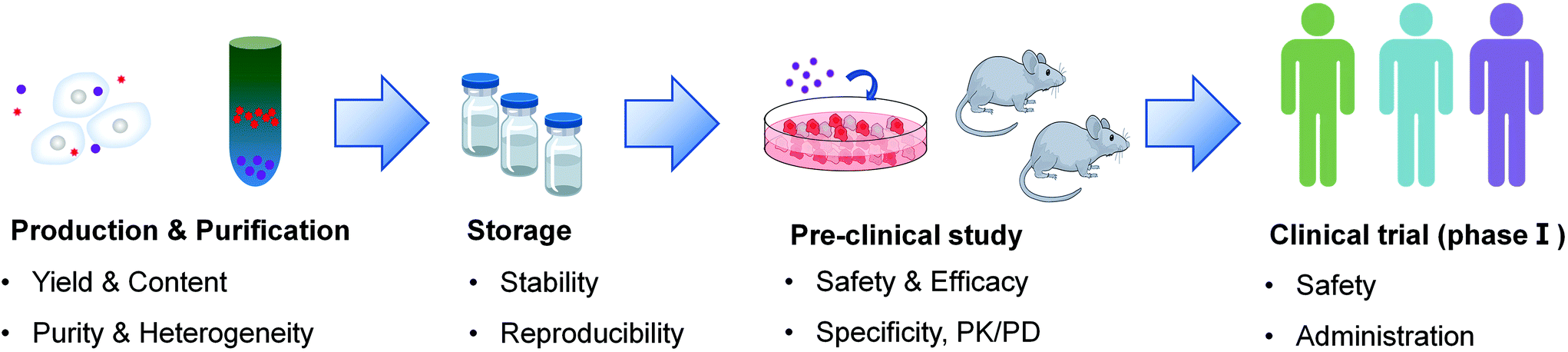 | ||
| Fig. 4 Main challenges in the clinical translation of EV-based therapeutics. | ||
5.1. Yield
In the actual process, a certain yield is needed to support its clinical application. To date, several factors are reported to influence the yield of EVs. Firstly, the external stimulus may affect the secretion of EVs. As reported by McNeill et al., the proliferation of CD34+ cells cultured on the type I collagen biomaterials was faster compared with that on fibronectin.77 This was attributed to the enhanced production of EVs containing miRNA-21 stimulated by collagen biomaterials. And the functional performance of CD34+ cells also improved, showing enhanced migration and angiogenic potential, which promoted the application of CD34+ cells in CABG surgery.Gupta et al. developed a protocol for producing mycobacterial extracellular vesicles (MEVs).78 They utilized the mechanism that the iron limitation would induce the enhanced release of MEVs in Mycobacterium tuberculosis and isolated the MEVs from the prepared iron-depleted defined medium. The purified MEVs were eight times higher than isolated from high iron conditions.
Besides, the modulation to the biogenesis of EVs may also cause an effect in their yield.15,34 For instance, Sung et al. reported that thrombin preconditioning could boost the biogenesis of EVs from mesenchymal stem cells with more cargo contents.79 They demonstrated that the mechanism mainly depends on the protease-activated receptor-1 (PAR-1) mediated signal pathways as this effect was inhibited by adding SCH79797, a PAR-1 signal antagonist. Thus, developing PAR-1-specific agonists might be a promising approach for EV yield improvement.
Moreover, a novel culture platform may also participate in the production of EVs. Based on the classical method of producing EVs in flask cultures, Watson et al. reported a hollow fiber bioreactor for producing EVs.80 Compared with the flask, the hollow fiber bioreactor reduced the confusion from the EVs of FBS. However, the cultural conditions may influence the metabolic signature of EVs, and the fiber bioreactor did not require fetal bovine serum (FBS), which may limit the application of a novel culture approach.81 Thus, the determination of cultivation conditions becomes extremely important. Patel et al. synthesized the expected culture scaffold for the cultivation of human dermal microvascular endothelial cells (HDMECs) via 3D-printing technology.82 The dynamic environment in the scaffold induced an enhanced amount of EVs from HDMECs, but the proportion of required EVs decreased and the total protein of each EV was also lost, which greatly reduced the therapeutic effect as their vascularization bioactivity. However, they employed an ethanol conditioning approach, which consequently enhanced the potency of EVs. This combination overcame the disadvantage of the bioreactor, that is, achieving yield at the expense of potency, which further promotes the application of bioreactors in the production of EVs.
Gao et al. developed another novel technology for EV production.83 The neutrophil-like cells (HL-60 cells) were directly disrupted via nitrogen cavitation (a physical force) and then self-assembly to nanovesicles. Compared with natural secretion, nitrogen cavitation contains similar structures and compositions, but the yield improved by 14 times higher. Then, they also mentioned that the nitrogen cavitation EVs contains less genetic content and subcellular organelles, which may lead to less heterogeneity and higher biocompatibility. Meanwhile, they utilized nitrogen cavitation EVs as a DDS in acute lung injury and sepsis, and firmly believed its wide application in medicine.
Hybridization with synthetic liposomes is another effective approach to improve the yield of EVs. The exosomes and small microvesicles with sizes below 200 nm are called sEVs, which serve as brilliant nanocarriers. However, the lack of modification flexibility and poor yield limited their further development. Thus, Rayamajhi et al. hybridized the sEVs with liposomes and demonstrated that the hybrid exosomes not only retained tumor-targeting properties and biocompatibility carried from macrophages but also obtained the advantages of the liposome, emerging with better drug release characteristic, greater colloidal stability and higher drug loading.84
5.2. Heterogeneity
Another challenge in the clinical translation of EVs is attributed to their heterogeneity. It is widely accepted that EVs are heterogeneous vesicles. The size, molecular compositions and biological functions of EVs are not only cell-type dependent but also can differ even when the exosomes originate from the same parental cells. Palma et al. demonstrated that breast cancer cells can secrete several types of exosomes, which differ in their size and CD44 content.85 Many studies have shown that different biomolecular factors including the physiological and pathological state of the parental cell, extracellular stimuli, and formation pathways can contribute to the heterogeneity of exosomes.86The miRNA content of EVs can differ even when the EVs are derived from the same tumor batch. While some miRNAs are highly expressed in most EVs, others are enriched only in specific subtypes of EVs. This phenomenon can be attributed to the fact that miRNAs are packaged into EVs through different mechanisms.87 Previous studies have shown that miRNAs in EVs represent a mixture of (a) highly expressed cellular miRNAs, which are passively incorporated into the EVs via an osmotic-like effect; (b) selectively secreted miRNAs, which are actively packaged into EVs based on the specific sequence of RNA molecules.88 For example, Pigati et al. found that about 66 percent of the released miRNAs are passively secreted through EVs depending on the amount of cytoplasmic miRNA, while 30 percent of exosomal miRNAs do not reflect the cellular profile, suggesting that they are selectively released.89
The protein of the EVs exhibited similar specialties to miRNA. For example, Wood and colleagues identified two subpopulations of EVs from B16F10 melanoma cells via sucrose density gradient centrifugation, termed LD-Exo and HD-Exo. Both of them enwrapped the same protein including Alix and TSG101. In addition to different species of proteins, LD-Exo contains unique proteins, actinin alpha 4 and cyclin Y, whereas the HD-Exo highly encases ephrin type-A receptor 2. They also discovered that the relative abundance of the same protein was not precisely in common.90 Besides, the EVs from apical versus basolateral sides of some cells could also vary much, which fed back in promoting and maintaining the polarization of cells.91 This effect was inhibited by Rab27a, which reduced the secretion of EVs via downgrading the biogenesis of ceramide. Another study reported that the Rab27a reduced the secretion of EVs via regulating intracellular compartments, whereas the 30–50 nm vesicles remained the same. This strongly indicated the different origins of EVs except from intracellular compartments, perhaps the plasma membrane.92 Meanwhile, previous studies demonstrated that the secretion of EVs relies on ESCRT-dependent or -independent sorting machinery, involving different molecules such as tetraspanins, which partly explains the different subtypes of EVs.21
Despite these findings, the majority of current studies use EVs as bulk isolates when evaluating their efficacy. The difficulties in separating specific exosomes include the lack of unique molecules to distinguish each EV subtype and appropriate isolation methods. Therefore, technological advances are urgently required to address this problem, which will help us to better understand exosome heterogeneity and accelerate the development of exosome-based therapeutics.
5.3. Stability
After the large-scale purification of EVs, the EV products require a suitable environment for storage to guarantee their stability. As an important issue in the clinical application of EVs, it requires more attention. Generally, a widely supported mode for storage of EVs is −80 °C,93 before which EVs are commonly resuspended in PBS.94 However, Crowe et al. reported that trehalose could improve the stability of EVs. A previous study demonstrated that trehalose is particularly effective in stabilizing dry membranes, phospholipid bilayers, and proteins.95 In this study, they suggested that trehalose limited the aggregation and fusion of beta-cell exosome-like vesicles (beta-ELV). And after repeated freezing and thawing, the integrity of beta-ELVs stored in trehalose was better than that in PBS. More importantly, the beta-ELVs stored in trehalose exhibited higher biological activity. All results suggested trehalose as a great cryoprotectant in cryogenic storage of clinical-grade EVs.96Although storage at −80 °C is a good choice, it may be limited by the cost and increase the difficulty of transportation. Charoenviriyakul et al. developed a promising storage method for EVs by lyophilization. They applied trehalose as a cryoprotectant to protect exosomes from the osmotic damage during lyophilization and stored the sample at room temperature after lyophilization. As shown by the results, the lyophilization had little effect on exosomes including the physical and biological characteristics.97 Due to the complexity of production, such as the different cell sources and isolation methods, optimizing the best storage conditions for each kind of EV therapeutic agent is particularly important, which still needs more research.
5.4. Specificity
Another major challenge hindering the utilization of EVs is the difficulty in ensuring delivery to their sites of therapeutic action while avoiding accumulation at off-target sites. Nonspecific delivery of EVs decreases the efficacy and may induce off-target effects. To date, several approaches to improve the specificity have been reported and achieved some effect.Ohno et al. developed a reliable method for building engineered cells.98 They developed an engineered HKE293 cell line via pDisplay vector transfection, which could secret GE11-positive exosomes. GE11 is a peptide that can specifically bind to EGFR (Epidermal Growth Factor Receptor). Thus, the peptide-positive exosomes derived from engineered HEK293 cells successfully delivered the therapeutic gene to EGFR+ breast cancer cells after systemic administration, causing tumor growth inhibition. Similarly, Alvarez-Erviti et al. utilized an engineered vector to transfect the DCs.99 The engineered DCs could produce EVs displaying Lamp2b, an exosomal membrane protein targeting the neuron-specific RVG peptide. Thus, the RVG-targeting EVs could specifically deliver the siRNA to the neurons in the brain and achieve gene therapy after systemic administration in mice.
However, screening new types of cells or reforming their characteristics might always be complicated and difficult. Thus, a universal and simple method is required. Membrane functionalization based on EVs is a superb choice. For example, Kooijmans et al. reported an approach by decorating EVs with recombinant phosphatidylserine-binding nanobodies (C1C2-nanobodies).100 After modification, EVs from RBCs and Neuro2A cells exhibited enhanced uptake in EGFR-overexpressing tumor cells compared to classical EVs, demonstrating the effect of C1C2-nanobodies in improving the tumor-targeting specificity. Other functional molecules, such as HA, achieved a similar effect.57
5.5. Safety
Finally, to achieve the goal of clinical translation, safety is undoubtedly a very important part of it. EVs are recognized as excellent delivery vehicles due to their high biocompatibility and low immunogenicity and toxicity. To date, EVs have been widely utilized in plenty of pre-clinical research and clinical research. However, many types of research studies acquiesced in the tolerance for EVs, whereas only part of them mentioned the security issues. Lately, a study particularly concentrated on the hepatotoxicity and immunogenicity of EVs.101In vitro, it found that the HepG2 cells did not exhibit significant influence both structurally and functionally even at a high exposure to EVs derived from Expi293F cells. It also demonstrated that the EVs did not induce the inflammatory response of recipient cells. And in vivo results supported that the relativity high dose of EVs did not induce toxicity and immune response in immune-sound mice. Meanwhile, although they originated from tumor cells, the EVs were proved to have no impact on the oncogenic or DNA damage pathways of HepG2 cells. Similar results were shown by Zhu et al.102 They intravenously and intraperitoneally dosed mice with HEK293T-derived EVs loaded with miR-199a-3p and chimeric proteins for 3 weeks. There was no significant toxicity and immune response of the therapy group as shown by hematology analysis and histopathological examination.Although these studies provide evidence for the immune-tolerance of EVs, further investigations are still desperately needed before clinical application, such as the effect of repeated injections, various routes of administration, and EVs from different cells with disparate bio-information.
6. Perspectives
Compared with existing synthetic carriers, EVs possess specific advantages, such as great biocompatibility and low immunogenicity and toxicity, as well as superior bio-stability. Meanwhile, EVs are rich resources as they can be easily obtained from body fluids including semen, urine, blood and bronchoalveolar lavage fluid. They can also be isolated from conditioned media. Thus, more scientific resources are devoted to this area, which extremely accelerates its development.Plenty of research focuses on the biological process of EVs, including their bio-genesis, transfer, biological effect and extinction. And they also provided multiple feasible approaches for the application of EVs in cancer diagnosis or therapy, such as modification with anti-tumor antigens for immunoregulation and enwrapping with therapeutic genes or drugs for tumor-targeting delivery. However, despite these present achievements, the clinical translation of EVs still faces several challenges.
First, the industrial production of EVs is still at an early stage, which urgently needs mass production and an isolation technology. Generally, a suitable industrial procedure may need to fulfill the following requirements: high yield, being simple and time-saving and reproducibility, which are exactly the shortcomings of existing methods. Next, several pre-clinical studies reported a series of approaches for the engineering of EVs. Among them, the functionalization of the EV membrane becomes a promising method. For example, the insertion of the polypeptide may improve the specificity of EVs including higher tumor-targeting ability or cellular uptake. However, it brings out the consideration that whether the insertion of exogenous molecules will induce unwanted changes of the EV initial functions, such as the enhanced immunogenicity and decreased stability, which asks for further investigation. Meanwhile, since the structure and effect of EVs are sensitive to the outside environment, the storage of EVs remains a great hurdle. Although cryopreservation seems harmless to EVs, it raises the cost of storage and transportation. Thus, lyophilization might be a great replacement owing to the low temperature requirement. The EV produced via lyophilization can even be stored at room temperature with little impact, which facilitates the commercialization of EV-based therapeutic agents. But the clinical data have yet to be supplemented. Finally, since EVs are heterogeneous particles, the standardization of the whole production process is urgent, either. It requires not only a unified procedure of production, isolation, characterization and storage, but also a recognized evaluation system for its safety and efficacy, as well as a guideline for the regimen of clinical administration. Moreover, each EV-based product must match a particular protocol, which exacerbates these difficulties. Considering these challenges, the clinical translation of EVs in cancer therapy still requires sustaining efforts. However, each breakthrough of these issues will greatly promote the clinical development of EVs, eventually leading to a revolution in cancer therapy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21805131 and 81673370) and the Shanghai Rising-Star Program (20QA1400900).Notes and references
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS.
- A. E. Sedgwick and C. D'Souza-Schorey, Traffic, 2018, 19, 319–327 CrossRef CAS.
- C. Théry, L. Zitvogel and S. Amigorena, Nat. Rev. Immunol., 2002, 2, 569–579 CrossRef.
- D. S. Chulpanova, K. V. Kitaeva, V. James, A. A. Rizvanov and V. V. Solovyeva, Front. Immunol., 2018, 9, 1534 CrossRef.
- N. P. Hessvik and A. Llorente, Cell. Mol. Life Sci., 2018, 75, 193–208 CrossRef CAS.
- Y. Zhang, Y. Liu, H. Liu and W. H. Tang, Cell Biosci., 2019, 9, 19 CrossRef.
- S. E. Andaloussi, I. Mäger, X. O. Breakefield and M. J. Wood, Nat. Rev. Drug Discovery, 2013, 12, 347–357 CrossRef.
- E. Willms, C. Cabañas, I. Mäger, M. J. Wood and P. Vader, Front. Immunol., 2018, 9, 738 CrossRef.
- R. Xu, A. Rai, M. Chen, W. Suwakulsiri, D. W. Greening and R. J. Simpson, Nat. Rev. Clin. Oncol., 2018, 15, 617–638 CrossRef CAS.
- K. Al-Nedawi, Nat. Cell Biol., 2008, 10, 619–624 CrossRef CAS.
- D. Di Vizio, J. Kim, M. H. Hager, M. Morello, W. Yang, C. J. Lafargue, L. D. True, M. A. Rubin, R. M. Adam and R. Beroukhim, Cancer Res., 2009, 69, 5601–5609 CrossRef CAS.
- D. Mazurov, L. Barbashova and A. Filatov, FEBS J., 2013, 280, 1200–1213 CrossRef CAS.
- A. Bobrie, M. Colombo, S. Krumeich, G. Raposo and C. Théry, J. Extracell. Vesicles, 2012, 1, 18397 CrossRef CAS.
- E. Faught, L. Henrickson and M. M. Vijayan, J. Endocrinol., 2017, 232, 237–246 CAS.
- F. Mobarrez, C. Sjövik, A. Soop, L. Hållström, C. Frostell, D. S. Pisetsky and H. Wallén, Platelets, 2015, 26, 486–490 CrossRef CAS.
- B. J. Goldie, M. D. Dun, M. Lin, N. D. Smith, N. M. Verrills, C. V. Dayas and M. J. Cairns, Nucleic Acids Res., 2014, 42, 9195–9208 CrossRef CAS.
- D. Choi, C. Spinelli, L. Montermini and J. Rak, Proteomics, 2019, 19, 1800169 CrossRef.
- E. R. Abels and X. O. Breakefield, Cell. Mol. Neurobiol., 2016, 301–312 CrossRef CAS.
- T. Skotland, K. Sandvig and A. Llorente, Prog. Lipid Res., 2017, 66, 30–41 CrossRef CAS.
- M. Record, M. Poirot and S. Silvente-Poirot, Biochimie, 2014, 96, 67–74 CrossRef CAS.
- G. Van Niel, G. d'Angelo and G. Raposo, Nat. Rev. Mol. Cell Biol., 2018, 19, 213–228 CrossRef CAS.
- D. N. Silachev, K. V. Goryunov, M. A. Shpilyuk, O. S. Beznoschenko, N. Y. Morozova, E. E. Kraevaya, V. A. Popkov, I. B. Pevzner, L. D. Zorova and E. A. Evtushenko, Cells, 2019, 8, 258 CrossRef CAS.
- Y. Qin, L. Wang, Z. Gao, G. Chen and C. Zhang, Sci. Rep., 2016, 6, 21961 CrossRef CAS.
- T. N. Lamichhane, S. Sokic, J. S. Schardt, R. S. Raiker, J. W. Lin and S. M. Jay, Tissue Eng., Part B, 2015, 21, 45–54 CrossRef CAS.
- P. D. Robbins, A. Dorronsoro and C. N. Booker, J. Clin. Invest., 2016, 126, 1173–1180 CrossRef.
- X. Zhang, H. Tu, Y. Yang, L. Fang, Q. Wu and J. Li, Stem Cells Int., 2017, 2017, 1758139 Search PubMed.
- A. Yekula, A. Yekula, K. Muralidharan, K. Kang, B. S. Carter and L. Balaj, Front. Immunol., 2019, 10, 3137 CrossRef CAS.
- R. Shah, T. Patel and J. E. Freedman, N. Engl. J. Med., 2018, 379, 958–966 CrossRef CAS.
- P. Li, M. Kaslan, S. H. Lee, J. Yao and Z. Gao, Theranostics, 2017, 7, 789–804 CrossRef CAS.
- S.-C. Guo, S.-C. Tao and H. Dawn, J. Extracell. Vesicles, 2018, 7, 1508271 CrossRef CAS.
- D. D. Taylor and S. Shah, Methods, 2015, 87, 3–10 CrossRef CAS.
- B. J. Benedikter, F. G. Bouwman, T. Vajen, A. C. Heinzmann, G. Grauls, E. C. Mariman, E. F. Wouters, P. H. Savelkoul, C. Lopez-Iglesias and R. R. Koenen, Sci. Rep., 2017, 7, 15297 CrossRef.
- M. Y. Konoshenko, E. A. Lekchnov, A. V. Vlassov and P. P. Laktionov, BioMed Res. Int., 2018, 2018, 8545347 Search PubMed.
- P. Duong, A. Chung, L. Bouchareychas and R. L. Raffai, PLoS One, 2019, 14, e0215324 CrossRef CAS.
- F. Yang, X. Liao, Y. Tian and G. Li, Biotechnol. J., 2017, 12, 1600699 CrossRef.
- H. Zhang, D. Freitas, H. S. Kim, K. Fabijanic, Z. Li, H. Chen, M. T. Mark, H. Molina, A. B. Martin and L. Bojmar, Nat. Cell Biol., 2018, 20, 332–343 CrossRef CAS.
- H. Shao, H. Im, C. M. Castro, X. Breakefield, R. Weissleder and H. Lee, Chem. Rev., 2018, 118, 1917–1950 CrossRef CAS.
- C. Gercel-Taylor, S. Atay, R. H. Tullis, M. Kesimer and D. D. Taylor, Anal. Biochem., 2012, 428, 44–53 CrossRef CAS.
- F. A. Coumans, A. R. Brisson, E. I. Buzas, F. Dignat-George, E. E. Drees, S. El-Andaloussi, C. Emanueli, A. Gasecka, A. Hendrix and A. F. Hill, Circ. Res., 2017, 120, 1632–1648 CrossRef CAS.
- A. Görgens, M. Bremer, R. Ferrer-Tur, F. Murke, T. Tertel, P. A. Horn, S. Thalmann, J. A. Welsh, C. Probst and C. Guerin, J. Extracell. Vesicles, 2019, 8, 1587567 CrossRef.
- W. Shen, K. Guo, G. B. Adkins, Q. Jiang, Y. Liu, S. Sedano, Y. Duan, W. Yan, S. E. Wang and K. Bergersen, Angew. Chem., Int. Ed., 2018, 57, 15675–15680 CrossRef CAS.
- Y. Kim, J. Jeon, S. Mejia, C. Q. Yao, V. Ignatchenko, J. O. Nyalwidhe, A. O. Gramolini, R. S. Lance, D. A. Troyer and R. R. Drake, Nat. Commun., 2016, 7, 11906 CrossRef CAS.
- L. Zhu, S. Kalimuthu, P. Gangadaran, J. M. Oh, H. W. Lee, S. H. Baek, S. Y. Jeong, S.-W. Lee, J. Lee and B.-C. Ahn, Theranostics, 2017, 7, 2732–2745 CrossRef CAS.
- W. M. Usman, T. C. Pham, Y. Y. Kwok, L. T. Vu, V. Ma, B. Peng, Y. S. Chan, L. Wei, S. M. Chin, A. Azad, A. B. He, A. Y. H. Leung, M. Yang, N. Shyh-Chang, W. C. Cho, J. Shi and M. T. N. Le, Nat. Commun., 2018, 9, 2359 CrossRef.
- K. O'brien, S. Khan, K. Gilligan, H. Zafar, P. Lalor, C. Glynn, C. O'Flatharta, H. Ingoldsby, P. Dockery and A. De Bhulbh, Oncogene, 2018, 37, 2137–2149 CrossRef.
- E. Erkan, D. Senfter, S. Madlener, G. Jungwirth, T. Ströbel, N. Saydam and O. Saydam, Cancer Gene Ther., 2017, 24, 38–44 CrossRef CAS.
- Q. Cheng, X. Shi, M. Han, G. Smbatyan, H. J. Lenz and Y. Zhang, J. Am. Chem. Soc., 2018, 140, 16413–16417 CrossRef CAS.
- Z. Yuan, K. K. Kolluri, K. H. Gowers and S. M. Janes, J. Extracell. Vesicles, 2017, 6, 1265291 CrossRef.
- Z. Yang, J. Shi, J. Xie, Y. Wang, J. Sun, T. Liu, Y. Zhao, X. Zhao, X. Wang, Y. Ma, V. Malkoc, C. Chiang, W. Deng, Y. Chen, Y. Fu, K. J. Kwak, Y. Fan, C. Kang, C. Yin, J. Rhee, P. Bertani, J. Otero, W. Lu, K. Yun, A. S. Lee, W. Jiang, L. Teng, B. Y. S. Kim and L. J. Lee, Nat. Biomed. Eng., 2020, 4, 69–83 CrossRef CAS.
- C. D. Hein, X.-M. Liu and D. Wang, Pharm. Res., 2008, 25, 2216–2230 CrossRef CAS.
- T. Smyth, K. Petrova, N. M. Payton, I. Persaud, J. S. Redzic, M. W. Graner, P. Smith-Jones and T. J. Anchordoquy, Bioconjugate Chem., 2014, 25, 1777–1784 CrossRef CAS.
- G. Jia, Y. Han, Y. An, Y. Ding, C. He, X. Wang and Q. Tang, Biomaterials, 2018, 178, 302–316 CrossRef CAS.
- S. Li, Y. Wu, F. Ding, J. Yang, J. Li, X. Gao, C. Zhang and J. Feng, Nanoscale, 2020, 12, 10854–10862 RSC.
- Y. Lin, J. Wu, W. Gu, Y. Huang, Z. Tong, L. Huang and J. Tan, Adv. Sci., 2018, 5, 1700611 CrossRef.
- M. Piffoux, A. K. A. Silva, C. Wilhelm, F. Gazeau and D. Tareste, ACS Nano, 2018, 12, 6830–6842 CrossRef CAS.
- L. Huang, N. Gu, X. E. Zhang and D. B. Wang, Adv. Funct. Mater., 2019, 29, 1807189 CrossRef.
- J. Liu, Z. Ye, M. Xiang, B. Chang, J. Cui, T. Ji, L. Zhao, Q. Li, Y. Deng and L. Xu, Biomaterials, 2019, 223, 119475 CrossRef CAS.
- K. O. Jung, H. Jo, J. H. Yu, S. S. Gambhir and G. Pratx, Biomaterials, 2018, 177, 139–148 CrossRef CAS.
- D. Ingato, J. A. Edson, M. Zakharian and Y. J. Kwon, ACS Nano, 2018, 12, 9568–9577 CrossRef CAS.
- S. Kamerkar, V. S. LeBleu, H. Sugimoto, S. Yang, C. F. Ruivo, S. A. Melo, J. J. Lee and R. Kalluri, Nature, 2017, 546, 498–503 CrossRef CAS.
- Y. Hong, G.-H. Nam, E. Koh, S. Jeon, G. B. Kim, C. Jeong, D.-H. Kim, Y. Yang and I.-S. Kim, Adv. Funct. Mater., 2018, 28, 1703074 CrossRef.
- D. Zhang, X. Qin, T. Wu, Q. Qiao, Q. Song and Z. Zhang, Biomaterials, 2019, 197, 220–228 CrossRef CAS.
- T. Yong, X. Zhang, N. Bie, H. Zhang, X. Zhang, F. Li, A. Hakeem, J. Hu, L. Gan, H. A. Santos and X. Yang, Nat. Commun., 2019, 10, 3838 CrossRef.
- R. J. C. Bose, S. Uday Kumar, Y. Zeng, R. Afjei, E. Robinson, K. Lau, A. Bermudez, F. Habte, S. J. Pitteri, R. Sinclair, J. K. Willmann, T. F. Massoud, S. S. Gambhir and R. Paulmurugan, ACS Nano, 2018, 12, 10817–10832 CrossRef.
- C. Théry, M. Boussac, P. Véron, P. Ricciardi-Castagnoli, G. Raposo, J. Garin and S. Amigorena, J. Immunol., 2001, 166, 7309–7318 CrossRef.
- F. Andre, N. Chaput, N. E. Schartz, C. Flament, N. Aubert, J. Bernard, F. Lemonnier, G. Raposo, B. Escudier, D. H. Hsu, T. Tursz, S. Amigorena, E. Angevin and L. Zitvogel, J. Immunol., 2004, 172, 2126–2136 CrossRef CAS.
- L. Zitvogel, A. Regnault, A. Lozier, J. Wolfers, C. Flament, D. Tenza, P. Ricciardi-Castagnoli, G. Raposo and S. Amigorena, Nat. Med., 1998, 4, 594–600 CrossRef CAS.
- M. A. Morse, J. Garst, T. Osada, S. Khan, A. Hobeika, T. M. Clay, N. Valente, R. Shreeniwas, M. A. Sutton, A. Delcayre, D. H. Hsu, J. B. Le Pecq and H. K. Lyerly, J. Transl. Med., 2005, 3, 9 CrossRef.
- B. Escudier, T. Dorval, N. Chaput, F. Andre, M. P. Caby, S. Novault, C. Flament, C. Leboulaire, C. Borg, S. Amigorena, C. Boccaccio, C. Bonnerot, O. Dhellin, M. Movassagh, S. Piperno, C. Robert, V. Serra, N. Valente, J. B. Le Pecq, A. Spatz, O. Lantz, T. Tursz, E. Angevin and L. Zitvogel, J. Transl. Med., 2005, 3, 10 CrossRef.
- S. Viaud, S. Ploix, V. Lapierre, C. Théry, P. H. Commere, D. Tramalloni, K. Gorrichon, P. Virault-Rocroy, T. Tursz, O. Lantz, L. Zitvogel and N. Chaput, J. Immunother., 2011, 34, 65–75 CrossRef.
- B. Besse, M. Charrier, V. Lapierre, E. Dansin, O. Lantz, D. Planchard, T. Le Chevalier, A. Livartoski, F. Barlesi, A. Laplanche, S. Ploix, N. Vimond, I. Peguillet, C. Thery, L. Lacroix, I. Zoernig, K. Dhodapkar, M. Dhodapkar, S. Viaud, J. C. Soria, K. S. Reiners, E. Pogge von Strandmann, F. Vely, S. Rusakiewicz, A. Eggermont, J. M. Pitt, L. Zitvogel and N. Chaput, OncoImmunology, 2016, 5, e1071008 CrossRef.
- S. Dai, D. Wei, Z. Wu, X. Zhou, X. Wei, H. Huang and G. Li, Mol. Ther., 2008, 16, 782–790 CrossRef CAS.
- J. Weber, V. K. Sondak, R. Scotland, R. Phillip, F. Wang, V. Rubio, T. B. Stuge, S. G. Groshen, C. Gee, G. G. Jeffery, S. Sian and P. P. Lee, Cancer, 2003, 97, 186–200 CrossRef CAS.
- P. Xu, K. Tang, J. Ma, H. Zhang, D. Wang, L. Zhu, J. Chen, K. Wei, J. Liu, H. Fang, L. Tang, Y. Zhang, J. Xie, Y. Liu, R. Meng, L. Liu, X. Dong, K. Yang, G. Wu, F. Ma and B. Huang, Cancer Immunol. Res., 2020, 8, 1193–1205 Search PubMed.
- Y. Gao, H. Zhang, N. Zhou, P. Xu, J. Wang, Y. Gao, X. Jin, X. Liang, J. Lv, Y. Zhang, K. Tang, J. Ma, H. Zhang, J. Xie, F. Yao, W. Tong, Y. Liu, X. Wang and B. Huang, Nat. Biomed. Eng., 2020, 4, 743–753 CrossRef CAS.
- J. Ma, Y. Zhang, K. Tang, H. Zhang, X. Yin, Y. Li, P. Xu, Y. Sun, R. Ma, T. Ji, J. Chen, S. Zhang, T. Zhang, S. Luo, Y. Jin, X. Luo, C. Li, H. Gong, Z. Long, J. Lu, Z. Hu, X. Cao, N. Wang, X. Yang and B. Huang, Cell Res., 2016, 26, 713–727 CrossRef CAS.
- B. McNeill, A. Ostojic, K. J. Rayner, M. Ruel and E. J. Suuronen, FASEB J., 2019, 33, 4166–4177 CrossRef CAS.
- S. Gupta and G. M. Rodriguez, J. Visualized Exp., 2019, e60359 CAS.
- D. K. Sung, S. I. Sung, S. Y. Ahn, Y. S. Chang and W. S. Park, Int. J. Mol. Sci., 2019, 20, 2899 CrossRef CAS.
- D. C. Watson, D. Bayik, A. Srivatsan, C. Bergamaschi, A. Valentin, G. Niu, J. Bear, M. Monninger, M. Sun and A. Morales-Kastresana, Biomaterials, 2016, 105, 195–205 CrossRef CAS.
- M. Palviainen, H. Saari, O. Kärkkäinen, J. Pekkinen, S. Auriola, M. Yliperttula, M. Puhka, K. Hanhineva and P. R.-M. Siljander, J. Extracell. Vesicles, 2019, 8, 1596669 CrossRef CAS.
- D. B. Patel, C. R. Luthers, M. J. Lerman, J. P. Fisher and S. M. Jay, Acta Biomater., 2019, 95, 236–244 CrossRef CAS.
- J. Gao, S. Wang and Z. Wang, Biomaterials, 2017, 135, 62–73 CrossRef CAS.
- S. Rayamajhi, T. D. T. Nguyen, R. Marasini and S. Aryal, Acta Biomater., 2019, 94, 482–494 CrossRef CAS.
- J. Palma, S. C. Yaddanapudi, L. Pigati, M. A. Havens, S. Jeong, G. A. Weiner, K. M. Weimer, B. Stern, M. L. Hastings and D. M. Duelli, Nucleic Acids Res., 2012, 40, 9125–9138 CrossRef CAS.
- R. Kalluri, J. Clin. Invest., 2016, 126, 1208–1215 CrossRef.
- R. Xu, A. Rai, M. Chen, W. Suwakulsiri, D. W. Greening and R. J. Simpson, Nat. Rev. Clin. Oncol., 2018, 15, 617–638 CrossRef CAS.
- B. N. Hannafon, Y. D. Trigoso, C. L. Calloway, Y. D. Zhao, D. H. Lum, A. L. Welm, Z. J. Zhao, K. E. Blick, W. C. Dooley and W. Q. Ding, Breast Cancer Res., 2016, 18, 90 CrossRef.
- L. Pigati, S. C. Yaddanapudi, R. Iyengar, D. J. Kim, S. A. Hearn, D. Danforth, M. L. Hastings and D. M. Duelli, PLoS One, 2010, 5, e13515 CrossRef.
- E. Willms, H. J. Johansson, I. Mager, Y. Lee, K. E. Blomberg, M. Sadik, A. Alaarg, C. I. Smith, J. Lehtio, S. El Andaloussi, M. J. Wood and P. Vader, Sci. Rep., 2016, 6, 22519 CrossRef CAS.
- A. R. Chin, W. Yan, M. Cao, X. Liu and S. E. Wang, J. Mammary Gland Biol. Neoplasia, 2018, 23, 165–176 CrossRef.
- A. Bobrie, M. Colombo, S. Krumeich, G. Raposo and C. Thery, J. Extracell. Vesicles, 2012, 1, 18397 CrossRef CAS.
- A. Jeyaram and S. M. Jay, AAPS J., 2017, 20, 1 Search PubMed.
- K. W. Witwer, E. I. Buzas, L. T. Bemis, A. Bora, C. Lasser, J. Lotvall, E. N. Nolte-‘t Hoen, M. G. Piper, S. Sivaraman, J. Skog, C. Thery, M. H. Wauben and F. Hochberg, J. Extracell. Vesicles, 2013, 2, 20360 CrossRef.
- J. H. Crowe, L. M. Crowe, J. F. Carpenter and C. Aurell Wistrom, Biochem. J., 1987, 242, 1–10 CrossRef CAS.
- S. Bosch, L. de Beaurepaire, M. Allard, M. Mosser, C. Heichette, D. Chretien, D. Jegou and J. M. Bach, Sci. Rep., 2016, 6, 36162 CrossRef CAS.
- C. Charoenviriyakul, Y. Takahashi, M. Nishikawa and Y. Takakura, Int. J. Pharm., 2018, 559, 427–428 CrossRef.
- S.-I. Ohno, M. Takanashi, K. Sudo, S. Ueda, A. Ishikawa, N. Matsuyama, K. Fujita, T. Mizutani, T. Ohgi and T. Ochiya, Mol. Ther., 2013, 21, 185–191 CrossRef CAS.
- L. Alvarez-Erviti, Y. Seow, H. Yin, C. Betts, S. Lakhal and M. J. Wood, Nat. Biotechnol., 2011, 29, 341–345 CrossRef CAS.
- S. A. Kooijmans, J. J. Gitz-Francois, R. M. Schiffelers and P. Vader, Nanoscale, 2018, 10, 2413–2426 RSC.
- A. F. Saleh, E. Lázaro-Ibáñez, M. A.-M. Forsgard, O. Shatnyeva, X. Osteikoetxea, F. Karlsson, N. Heath, M. Ingelsten, J. Rose and J. Harris, Nanoscale, 2019, 11, 6990–7001 RSC.
- X. Zhu, M. Badawi, S. Pomeroy, D. S. Sutaria, Z. Xie, A. Baek, J. Jiang, O. A. Elgamal, X. Mo, K. Perle, J. Chalmers, T. D. Schmittgen and M. A. Phelps, J. Extracell. Vesicles, 2017, 6, 1324730 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |