
A simple self-adjuvanting biomimetic nanovaccine self-assembled with the conjugate of phospholipids and nucleotides can induce a strong cancer immunotherapeutic effect†
Dan
Liu
a,
Jiale
Liu
a,
Bing
Ma
a,
Bo
Deng
a,
Xigang
Leng
a,
Deling
Kong
 ab and
Lanxia
Liu
*a
ab and
Lanxia
Liu
*a
aThe Tianjin Key Laboratory of Biomaterials, Institute of Biomedical Engineering, Peking Union Medical College & Chinese Academy of Medical Sciences, Tianjin 300192, China. E-mail: liulanxiabme@163.com; Fax: +86 (22) 8789-1191; Tel: +86 (22) 8789-1191
bCollege of Life Science, Nankai University, Tianjin 300071, China
First published on 11th September 2020
Abstract
Biomimetic nanoparticles have potential applications in many fields due to their favorable properties. Here, we developed a self-adjuvanting biomimetic anti-tumor nanovaccine, which was self-assembled with an amphiphilic conjugate synthesized with the phospholipids of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and hydrophilic Toll-like receptor (TLR9) agonist CpG ODN. The nanovaccine could not only provide effective initial antigen stimulation and sustained long-term antigen supply with a controlled release, but also induce antigen cross-presentation via the MHC-I pathway initiating CD8+ T-cell responses. Moreover, the dense nucleotide shell around the nanovaccine could promote antigen endocytosis via various receptor-mediated pathways into dendritic cells. CpG ODN interacted with TLR9 triggering the cytokine secretion of TNF-α and IL-10, which further boosted the anti-tumor humoral and cellular immune responses, which led to a significant tumor suppressive effect and remarkable survival prolongation. So, this nanovaccine self-assembled with phospholipid–nucleotide amphiphiles can serve as a safe, simple and efficient approach for anti-tumor immunotherapy.
1. Introduction
Nanovaccines have been investigated in recent years as an emerging field in cancer immunotherapy owing to their ability to cross biological barriers, prolong circulation times, and induce an enhanced long-lasting protective immune effect.1–3 Generally, nanovaccine carriers are required to deliver antigens and/or adjuvants to overcome intrinsic difficulties of functional cargos and trigger delightful immune responses.4–6 Many studies reported that the co-delivery of antigens and adjuvants of CpG ODN, whether encapsulated in or located outside the nanocarriers, could promote strong immune responses and enhance the anti-tumor effect.7,8 Recently, it has been reported that nucleic acids (CpG ODN) arranged in a dense spherical spatial form (SNAs) on a core made of gold or other polymers, can endow them with unique properties, such as rapid uptake through a receptor-mediated mechanism and efficient protection against nuclease degradation, and significantly enhance the immune response.9–12 However, the preparation of popular polymeric nanocarriers is complicated and traditional core polymers have potential physiological toxicity.6,13–16Biomimetic NPs to mimic the chemical and structural features of biological systems possess distinct properties, including favorable biocompatibility, extended circulation and targeted drug delivery. Here, we report a simple self-adjuvanting biomimetic nanovaccine (NaV) self-assembled with the conjugate of phospholipids and nucleotides, which had a dense nucleotide shell similar to SNAs. In this study, hydrophobic biomimetic phospholipids of DOPE and hydrophilic adjuvanting Toll-like receptor (TLR9) agonist CpG ODN were exploited and covalently linked together. The resultant amphiphilic molecules similar to the phospholipid bilayer of the cell membrane self-assembled forming nanocapsules. So, the nanovaccine mimicked both the chemical and structural characteristics of cells displaying good biocompatibility. Furthermore, CpG ODN not only as adjuvants enhanced the immune responses, but also the dense nucleotide shell formed with CpG ODN promoted antigen endocytosis via various receptor-mediated pathways and subsequently boosted the anti-tumor humoral and cellular immune responses. Therefore, the biomimetic nanovaccine offered a simple, safe and effective strategy for cancer treatment.
2. Materials and methods
2.1 Preparation of self-adjuvanting biomimetic nanovaccines
The preparation of self-adjuvanting biomimetic nanovaccines involved three steps. Briefly, DOPE (Sigma-Aldrich Co., USA) was pyridyldithiol-activated by 3-(2-pyridyldithio) propionic acid N-hydroxysuccinimide ester (SPDP). Then, the activated DOPE was conjugated with thiol terminated CpG ODN (Sangon Biotech Ltd, China) via a disulfide bond in the presence of a phase transfer catalyst. Finally, the resulted amphiphiles of DOPE-S-S-CpG ODN and ovalbumin (OVA, a model antigen, Sigma-Aldrich Co., USA) solutions were mixed and stirred at 25 °C for the formation of self-adjuvanting biomimetic nanovaccines (named NaVs) with self-assembly. The NaVs were washed with distilled water and lyophilized.2.2 Characterization of DOPE-S-S-CpG ODN amphiphiles and NaVs
The conjugate of DOPE-S-S-CpG ODN was confirmed by agarose gel electrophoresis. The particle sizes and zeta potential of the NaVs were analyzed using Zetasizer Nano ZP (Malvern Instruments Ltd, UK). The redox-responsive properties of the NaVs were studied through observing the changes in the size after reaction with excess DL-dithiothreitol (DTT). To investigate the stability of the NaVs, the size of the NaVs was measured every five days until the nanoparticles aggregated. The morphology of the NaVs was observed using transmission electron microscopy (TEM) (Tecnai-F20, FEI, the Netherlands).The OVA loading capacity of the NaVs was determined using an Enhanced-BCA Protein Assay Kit. To evaluate the release behavior of antigens from the NaVs in vitro, 5 mg of lyophilized NaV powder was dissolved with 1 mL of phosphate buffered saline (PBS, pH7.4) and placed in a dialysis bag (molecular weight: 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) which was immersed in 10 ml of PBS at 37 °C, and the OVA concentration in dialysate was measured with BCA kits at regular intervals.
000 Da) which was immersed in 10 ml of PBS at 37 °C, and the OVA concentration in dialysate was measured with BCA kits at regular intervals.
2.3 Immunization studies in vitro
2.4 Immunization studies in vivo
To further evaluate the anti-tumor effect of the NaVs, the length and width of mouse tumors were recorded with digital vernier calipers every two days, while observing the mental state of the mice. Tumor volume (mm3) was calculated according to the following formula: (length × width2)/2. Two days after the last immunization, the mice were sacrificed and their peripheral blood, tumors and spleens were collected immediately for the following immunological experiments. The solid tumor tissues collected from tumor-bearing mice were stained with hematoxylin and eosin (H&E) and TUNEL apoptosis kits (Beyotime Biotechnology, China) to observe the influence of the NaVs on the necrosis and apoptosis of tumor cells.
2.5 Statistical analysis
Data were expressed as mean ± standard deviation (SD). The differences were assessed using ANOVA or the Student's t-test and the Tukey's post-test. P value not more than 0.05 was considered significant.3. Results and discussion
3.1 NaV preparation and characterization studies
DOPE and CpG ODN were covalently linked using SPDP as the crosslinking agent. The successful synthesis of DOPE-S-S-CpG ODN was confirmed by agarose gel electrophoresis (Fig. S1†). The resulting DOPE-S-S-CpG ODN and OVA (as model antigen) self-assembled forming NaVs. The morphology of the NaVs was characterized by TEM and the image showed that the formulations were uniform nanocapsules (Fig. 1A). The diameter of the NaVs determined with dynamic light scattering (DLS) was 192.4 ± 0.66 nm, while that of the blank nanocapsule without OVA (named SNCs) was 138.2 ± 0.90 nm (Fig. 1B and Table S1†). The size of the NaVs remained stable in PBS solution for 45 days approximately (Fig. 1C). The loading capacity of OVA in NaVs determined by an Enhanced-BCA Protein Assay Kit was (26.4 ± 0.77)%. OVA had a burst release of 30% from the NaVs during the first two days under conditions that mimic the intracellular reducing environment (4 mM DTT in 7.4 pH buffer) followed by a controlled release reaching about 65% in 25 days, which was faster than the release behavior of OVA (44.99 ± 1.69)% under conditions without DTT (Fig. 1D), indicating that the NaVs did possess redox-responsive properties allowing antigens’ quick release after uptake into cells, and the nanovaccines could provide strong initial antigen stimuli and long-term sustained antigen exposure to immune cells, which was important for eliciting powerful immune responses.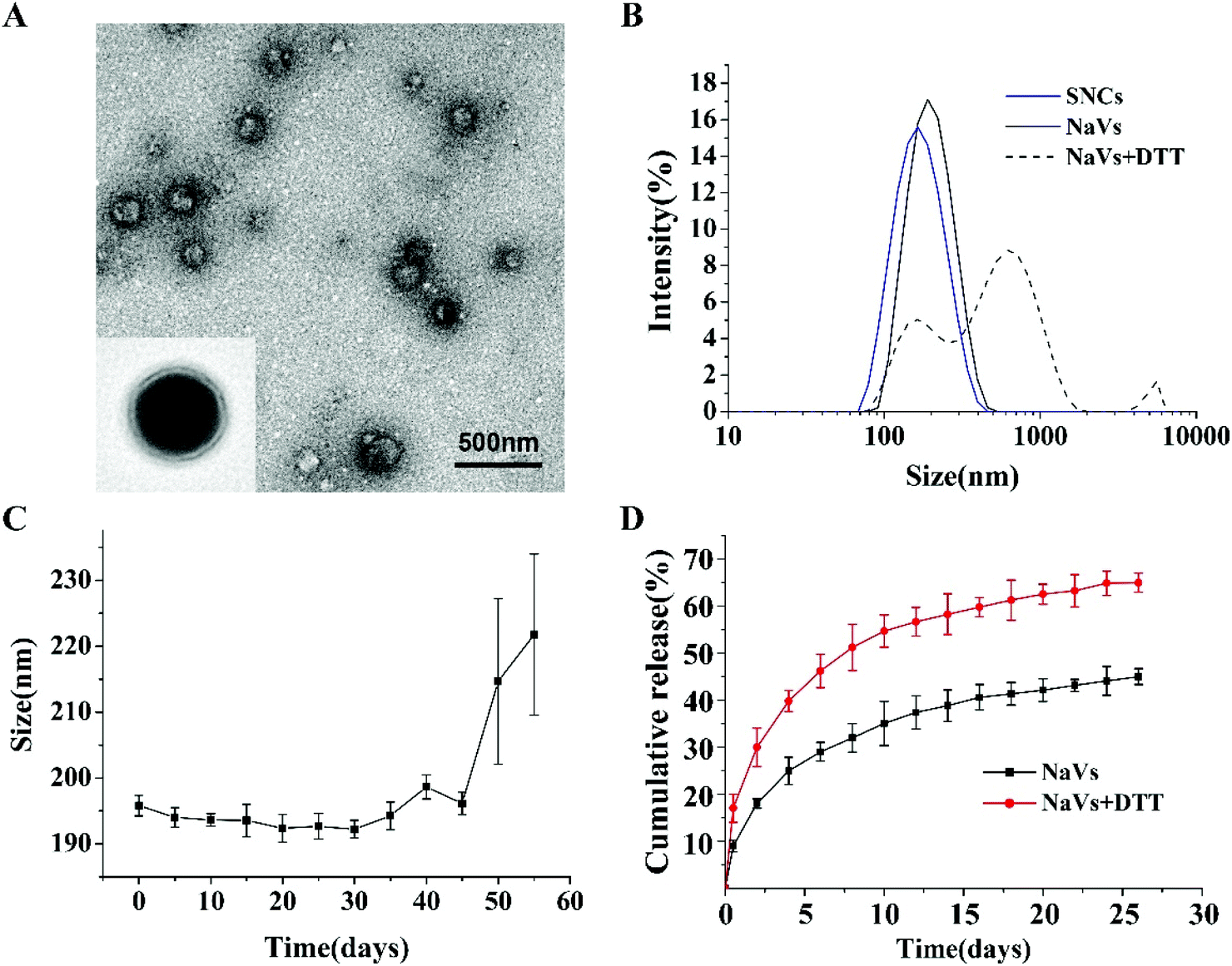 | ||
| Fig. 1 (A) Typical TEM images of NaVs. (B) The particle distribution of different formations, and (C) stability study of NaVs in vitro, and (D) in vitro OVA release profile from NPs with/without the DTT reaction in 7.4 pH buffer. | ||
3.2 Immunization studies in vitro
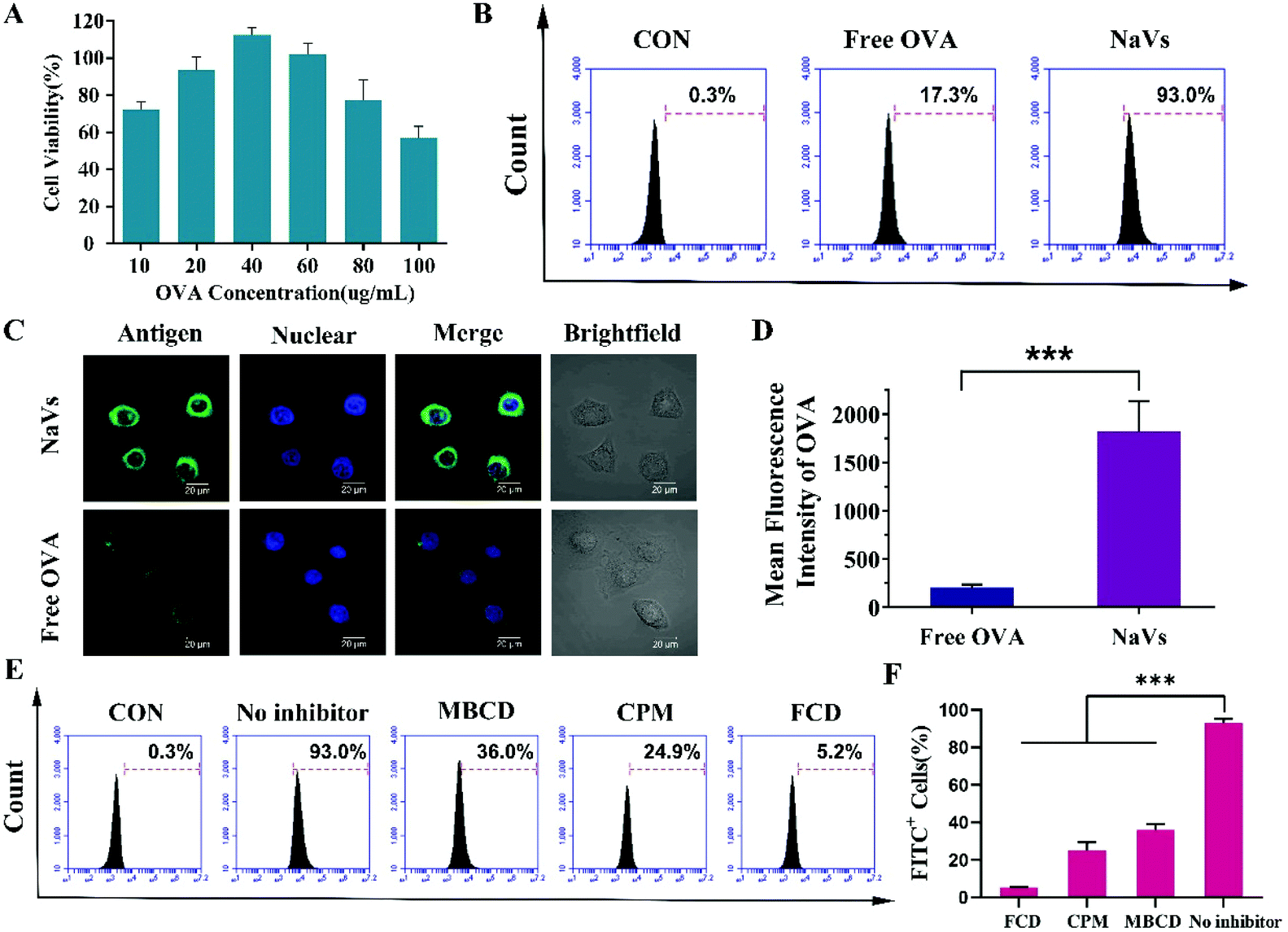 | ||
| Fig. 2 Studies on cytotoxicity and cellular uptake pathways of nanoparticles. (A) Cell viability at different concentrations of OVA. (B) Representative flow cytometry analysis of the cellular uptake of NaVs and free OVA. (C) CLSM images of BMDCs after incubation with free FITC-OVA and NaVs and (D) representative image of the mean fluorescence intensity analysis of OVA ingested by cells, analyzed using ImageJ software. (E) A representative flow cytometry profile of the nanoparticle uptake by DCs with different inhibitors, and (F) representative fluorescence intensity histogram. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Although Chad A. Mirkin et al. reported that high-density oligonucleotides on the surface of nanoparticles could bind scavenger receptors on the surface of macrophages and promote nanoparticle uptake into macrophages,12 in an attempt to further explore the endocytic pathways and investigate the mechanism of nanoparticle uptake in DCs that is important for initiating immune responses for vaccines, we pretreated BMDCs with methyl-β-cyclodextrin (MBCD), chlorpromazine (CPM) and fucoidan (FCD), respectively. MBCD can remove cholesterol from the cell membrane effectively inhibiting caveolin-dependent endocytosis, CPM can block the formation of clathrin-coated pits, while FCD is a pharmacological inhibitor of class A scavenger receptor (SR-A).17–20 As shown in Fig. 2E and F, compared to untreated cells, FCD led to a drastic reduction of the antigen uptake of the NaVs (dropped up to 5.2%), MBCD reduced the uptake of cells to 36.0%, while CPM caused an uptake decrease to 24.9%. All the data suggested that the NaVs internalized into BMDCs through multiple receptor-mediated endocytosis pathways, but mainly mediated by SR-A. Altogether, the NaVs constructed with the conjugate of adjuvant CpG ODN and biomimetic material DOPE having a “3D” structure could load antigens in the cavity of nanocapsules as vaccine delivery nanocarriers, and the high-density nucleotides on the surface of nanocapsules could bind the various receptors on the surface of BMDCs and promote antigen endocytosis circumventing the drawbacks of free antigens, which in turn facilitated the CpG ODN interaction with the TLR9 receptor in endosomes and activated antigen presenting cells.21–27
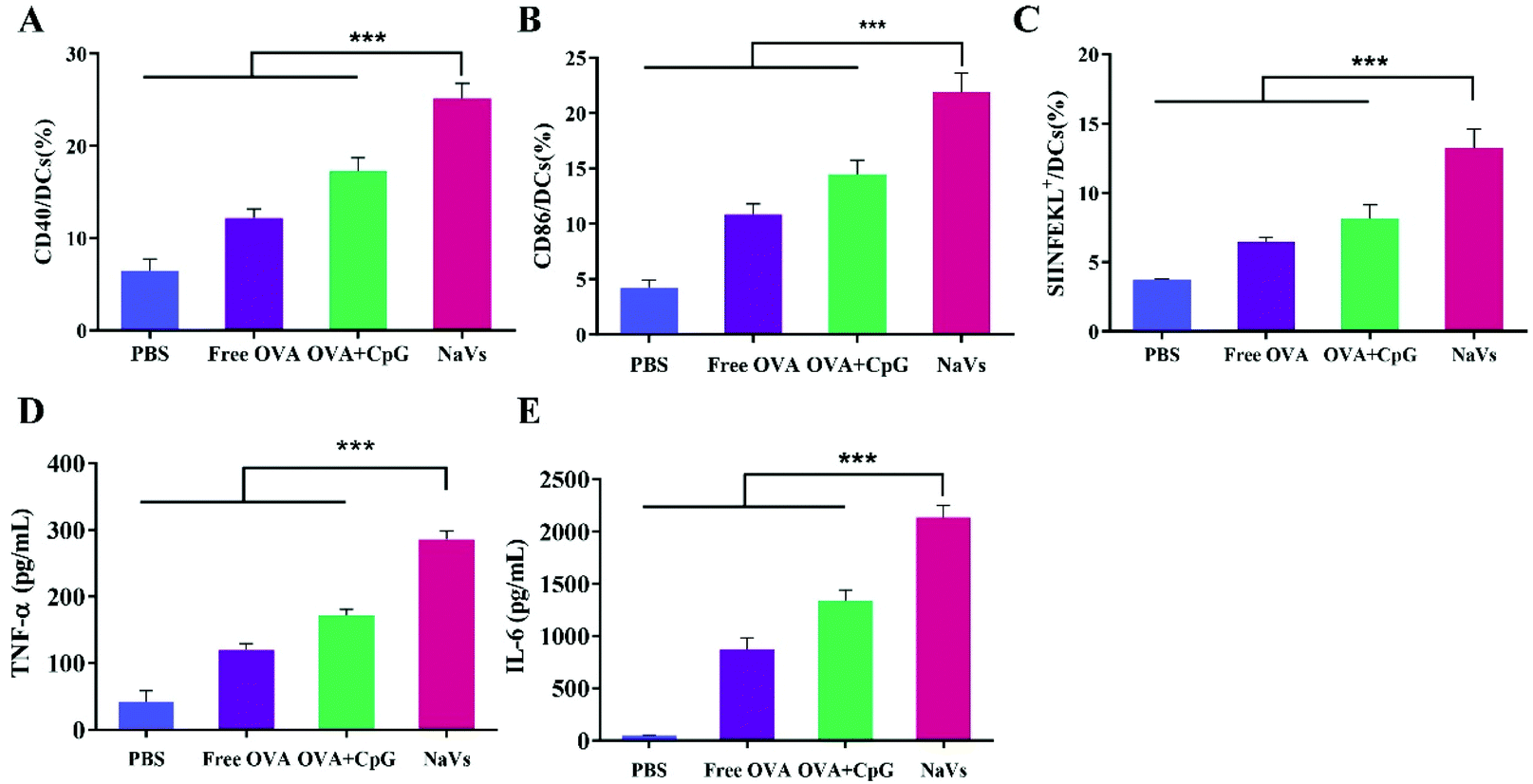 | ||
| Fig. 3 Effects of different components on the antigen cross-presentation and activation of BMDCs. Representative flow cytometry images of (A) CD40+, (B) CD86+ and (C) SIINFEKL+ cells. Expression levels of (D) TNF-α and (E) IL-6 in supernatants were determined with ELISA assays, after BMDCs were treated with PBS, free OVA, OVA + CpG and NaVs. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
3.3 Immunization studies in vivo
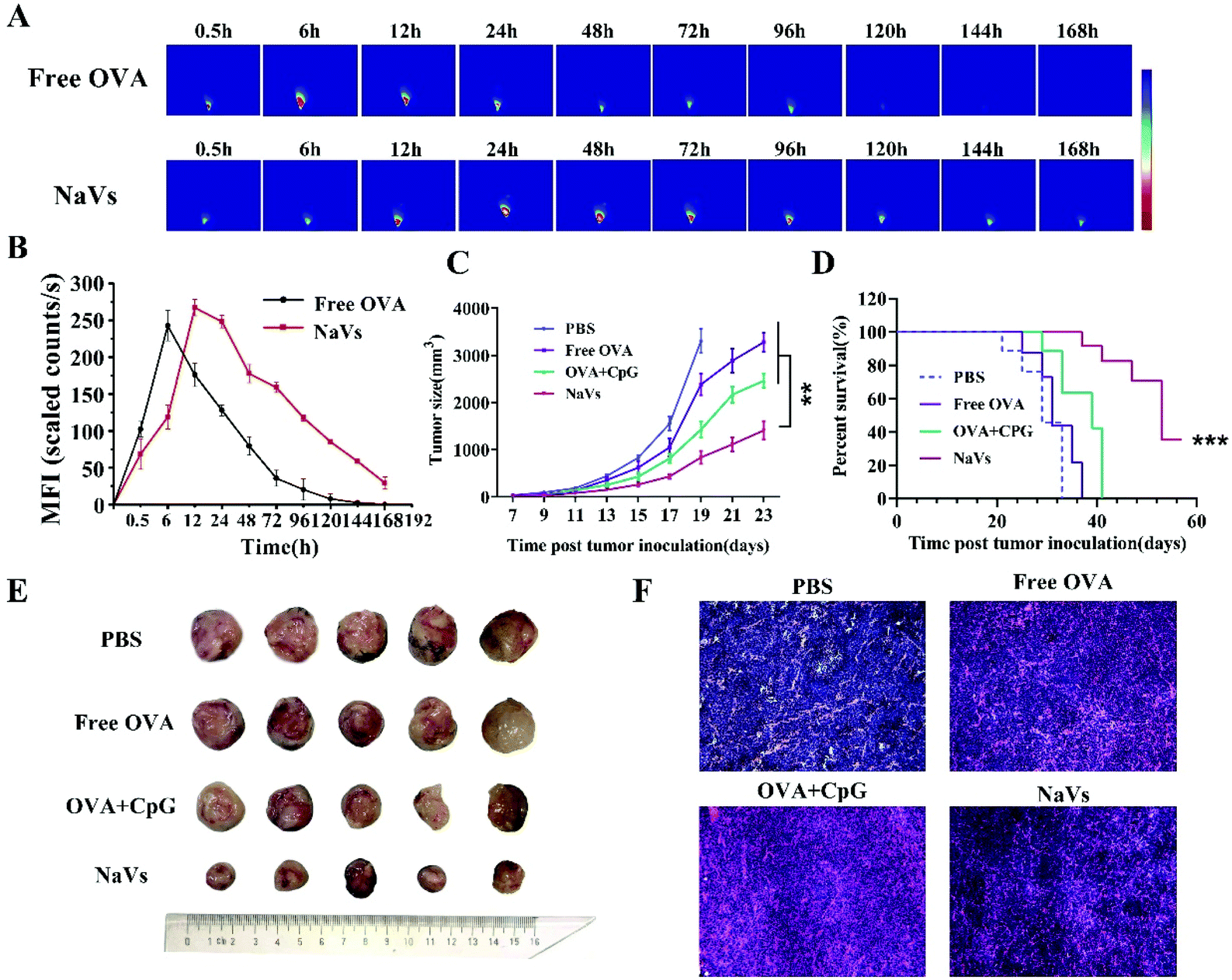 | ||
| Fig. 4 (A) Antigen release behavior of NaVs at the injection site. Representative fluorescence images of different formulations at different points in time after injection. (B) Average fluorescence intensity at the injected site within a predetermined time period. (C) Average tumor growth curves were recorded and plotted. (D) Survival of mice treated with PBS, free OVA, OVA + CpG or NaVs. (E) Tumors harvested from tumor-bearing mice after different component treatments, and (F) H&E staining of tumor tissues obtained from mice immunized with different formulations. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
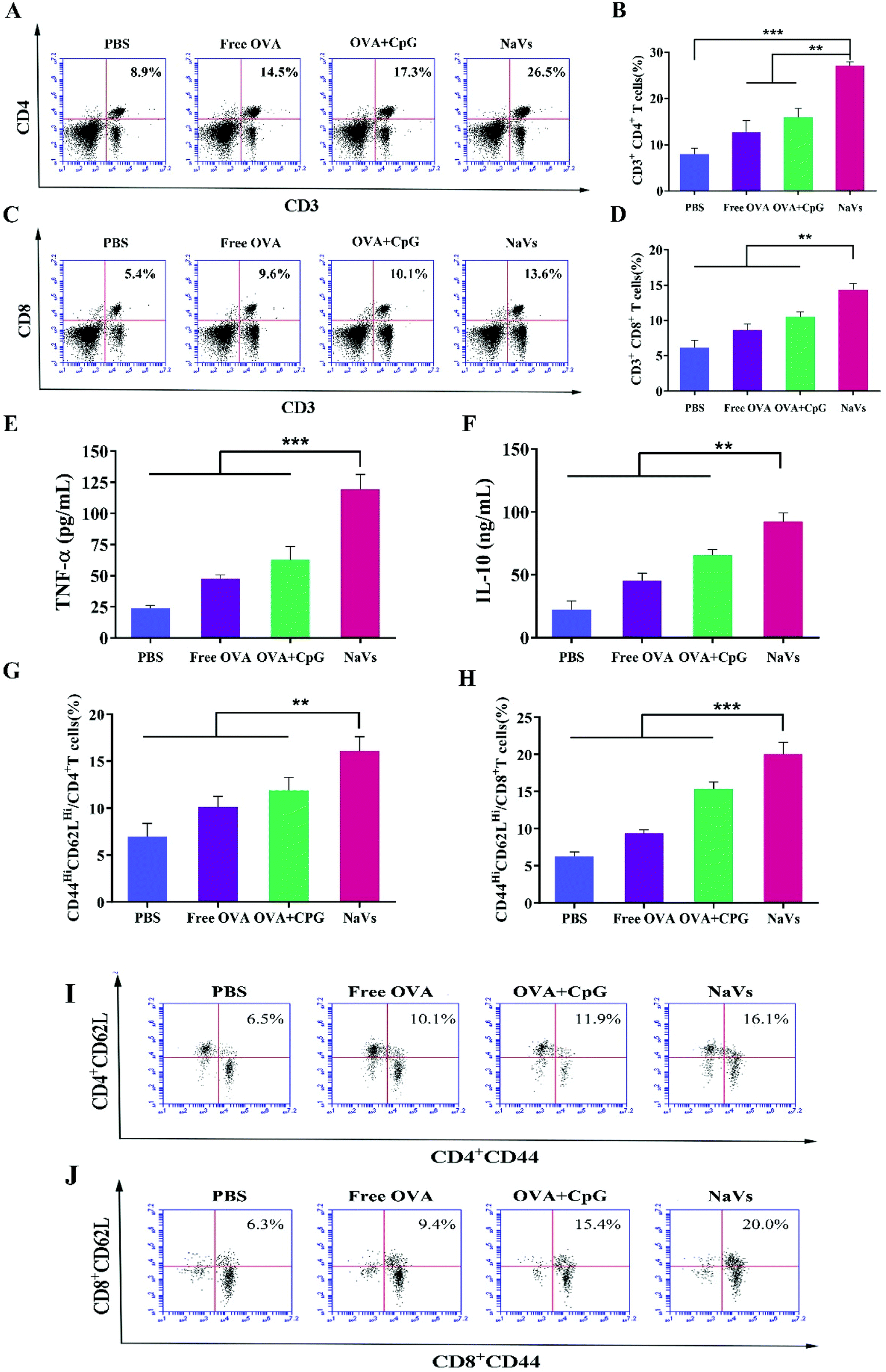 | ||
| Fig. 5 T immune cell activation and representative FACS plots. (A) Representative FACS plots and (B) percentage of CD3+CD4+ T cells and (C and D) CD3+CD8+ T cells were plotted for mice of different groups, followed by the histograms of (E) TNF-α and (F) IL-10. The type and proportion of memory T cells in splenocytes after stimulation were detected and analyzed, and the proportion of central memory T cells in (G) CD4+ T cell population or (H) CD8+ T cell population and their representative FACS plots are shown. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Furthermore, the cytokine levels of the supernatant of splenocytes after antigen re-stimulation in vitro were detected. As shown in Fig. 5E, the NaVs increased the cytokine level of TNF-α approximately 4 times compared with the PBS group, which is consistent with the in vitro results. The NaVs also significantly elevated the level of cytokine IL-10 (Fig. 5F). IL-10 as an anti-inflammatory factor can specifically limit the Th17 inflammation to eliminate the protumor environment and promote Th1-type anti-tumor immunity to recognize and eliminate tumors cells, and hence inhibit the tumor development and metastasis.37,38 It can also activate NK cells and CD8+T cells directly or indirectly to enhance anti-tumor effects and contribute to the formation of immune memory.39 As stated in the previous study, here the cytokines of TNF-α and IL-10 were upregulated, mainly mediated by NF-κB and AP-1 activation through mitogen-activated protein kinase (MAPK) and nuclear factor kappa-B inducing kinase-IkB kinase-IkB (NIK-IKK-IkB) pathways which resulted from TLR-9 receptor binding with dense CpG ODNs after the NaVs were internalized into endosomes.5,40 Altogether, we conclude that the NaVs could enhance both humoral and cellular immune responses.
4. Conclusions
In summary, as shown in Fig. 6, we developed a self-adjuvanting biomimetic nanovaccine self-assembled with the amphiphilic conjugate synthesized with hydrophobic phospholipids and hydrophilic TLR9 agonist CpG ODN. The nanovaccine with a 3D structure could load antigens in the cavity, provide effective initial immune stimulation and sustained long-term antigen supply, and promote antigen cross-presentation via the MHC-I pathway initiating CD8+ T-cell responses. Moreover, the dense nucleotide shell around the nanovaccine could promote antigen endocytosis via various receptor-mediated pathways into dendritic cells. The CpG ODN interaction with TLR9 triggering the cytokine secretion of TNF-α and IL-10 further boosted the anti-tumor humoral and cellular immune responses as well as generated effective immune memory, which led to a significant tumor suppressive effect and remarkable survival prolongation. So, the self-adjuvanting biomimetic nanovaccine can serve as a great promising immunotherapeutic approach for tumor treatment. Moreover, the method reported here could be applied to produce various types of nanovaccines for the treatment of infectious diseases though loading different antigens in the cavity of the capsule, or to prepare siRNA nanocarriers for gene therapy through replacing the CpG ODN shell around the nanovaccine with siRNA.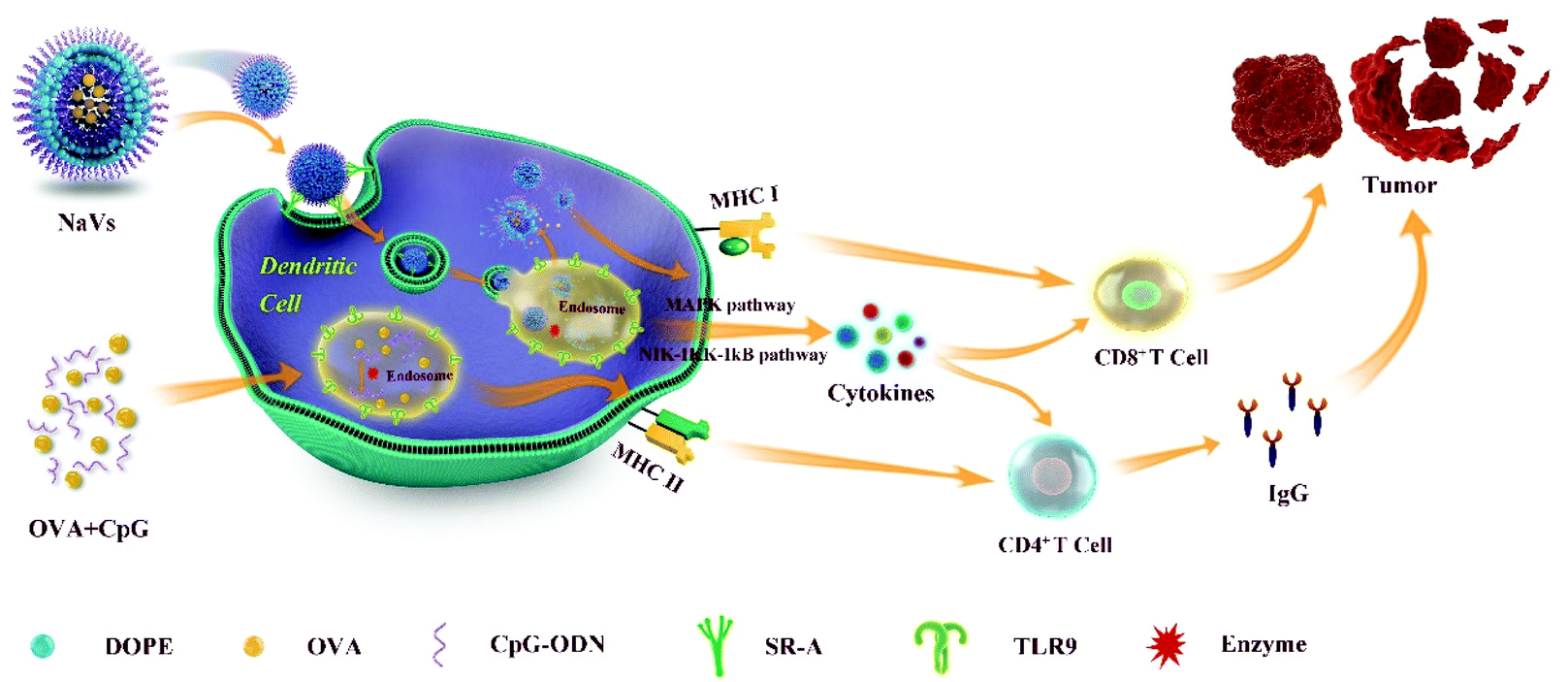 | ||
| Fig. 6 Illustration of the NaVs anti-tumor immune effect mechanism. | ||
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Natural Science Foundation of China (No. 31870920), CAMS Innovation Fund for Health and Longevity Pilot Project (Youth Award Program, 2019-RC-HL-015) and CAMS Innovation Fund for Medical Sciences (CAMS-I2M-3-004).References
- S. Acharya and S. K. Sahoo, Adv. Drug Delivery Rev., 2011, 63, 170–183 CrossRef CAS.
- K. Shao, S. Singha, X. Clemente-Casares, S. Tsai, Y. Yang and P. Santamaria, ACS Nano, 2015, 9, 16–30 CrossRef CAS.
- J. Zhou, A. V. Kroll, M. Holay, R. H. Fang and L. Zhang, Adv. Mater., 2019, e1901255, DOI:10.1002/adma.201901255.
- G. Zhu, F. Zhang, Q. Ni, G. Niu and X. Chen, ACS Nano, 2017, 11, 2387–2392 CrossRef CAS.
- M. Luo, H. Wang, Z. Wang, H. Cai, Z. Lu, Y. Li, M. Du, G. Huang, C. Wang, X. Chen, M. R. Porembka, J. Lea, A. E. Frankel, Y.-X. Fu, Z. J. Chen and J. Gao, Nat. Nanotechnol., 2017, 12, 648–654 CrossRef CAS.
- S. Singha, K. Shao, K. K. Ellestad, Y. Yang and P. Santamaria, ACS Nano, 2018, 12, 10621–10635 CrossRef CAS.
- T. Kang, Y. Huang, Q. Zhu, H. Cheng, Y. Pei, J. Feng, M. Xu, G. Jiang, Q. Song, T. Jiang, H. Chen, X. Gao and J. Chen, Biomaterials, 2018, 164, 80–97 CrossRef CAS.
- Y. Hu, L. Lin, J. Chen, A. Maruyama, H. Tian and X. Chen, Biomaterials, 2020, 252, 120114 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS.
- C. A. Mirkin and S. H. Petrosko, Clin. Chem., 2018, 64, 971–972 CrossRef CAS.
- J. R. Ferrer, A. J. Sinegra, D. Ivancic, X. Y. Yeap, L. Qiu, J.-J. Wang, Z. J. Zhang, J. A. Wertheim and C. A. Mirkin, ACS Nano, 2020, 14, 1682–1693 CrossRef CAS.
- C. H. J. Choi, L. Hao, S. P. Narayan, E. Auyeung and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7625–7630 CrossRef CAS.
- Y. Wang, L. Miao, A. Satterlee and L. Huang, Adv. Drug Delivery Rev., 2015, 87, 68–80 CrossRef CAS.
- L. Giodini, F. L. Re, D. Campagnol, E. Marangon, B. Posocco, E. Dreussi and G. Toffoli, Nanomedicine, 2017, 13, 583–599 CrossRef CAS.
- Y. Xin, M. Huang, W. W. Guo, Q. Huang, L. Z. Zhang and G. Jiang, Mol. Cancer, 2017, 16, 134 CrossRef.
- D. Shi, N. M. Bedford and H.-S. Cho, Small, 2011, 7, 2549–2567 CrossRef CAS.
- S. De Koker, B. G. De Geest, S. K. Singh, R. De Rycke, T. Naessens, Y. Van Kooyk, J. Demeester, S. C. De Smedt and J. Grooten, Angew. Chem., Int. Ed., 2009, 48, 8485–8489 CrossRef CAS.
- P. C. Patel, D. A. Giljohann, W. L. Daniel, D. Zheng, A. E. Prigodich and C. A. Mirkin, Bioconjugate Chem., 2010, 21, 2250–2256 CrossRef CAS.
- J. Rejman, A. Bragonzi and M. Conese, Mol. Ther., 2005, 12, 468–474 CrossRef CAS.
- D. A. Kuhn, D. Vanhecke, B. Michen, F. Blank, P. Gehr, A. Petri-Fink and B. Rothen-Rutishauser, Beilstein J. Nanotechnol., 2014, 5, 1625–1636 CrossRef.
- N. Hanagata, Int. J. Nanomed., 2017, 12, 515–531 CrossRef CAS.
- Y. M. Murad and T. M. Clay, Biodrugs, 2009, 23, 361–375 CrossRef CAS.
- H. Hemmi, O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda and S. Akira, Nature, 2000, 408, 740–745 CrossRef CAS.
- R. Kuai, X. Sun, W. Yuan, L. J. Ochyl, Y. Xu, A. Hassani Najafabadi, L. Scheetz, M.-Z. Yu, I. Balwani, A. Schwendeman and J. J. Moon, J. Controlled Release, 2018, 282, 131–139 CrossRef CAS.
- G. N. Barber, Immunol. Rev., 2011, 243, 99–108 CrossRef CAS.
- F. Takeshita, I. Gursel, K. J. Ishii, K. Suzuki, M. Gursel and D. M. Klinman, Semin. Immunol., 2004, 16, 17–22 CrossRef CAS.
- J. Yue, R. M. Pallares, L. E. Cole, E. E. Coughlin, C. A. Mirkin, A. Lee and T. W. Odom, ACS Appl. Mater. Interfaces, 2018, 10, 21920–21926 CrossRef CAS.
- O. P. Joffre, E. Segura, A. Savina and S. Amigorena, Nat. Rev. Immunol., 2012, 12, 557–569 CrossRef CAS.
- A. Gardner and B. Ruffell, Trends Immunol., 2016, 37, 855–865 CrossRef CAS.
- S. I. Grivennikov and M. Karin, Ann. Rheum. Dis., 2011, 70(Suppl 1), i104–i108 CrossRef CAS.
- S. A. Jones and B. J. Jenkins, Nat. Rev. Immunol., 2018, 18, 773–789 CrossRef CAS.
- F. Balkwill, Cancer Metastasis Rev., 2006, 25, 409–416 CrossRef CAS.
- F. Balkwill, Nat. Rev. Cancer, 2009, 9, 361–371 CrossRef CAS.
- J. Liu, X. Liu, Y. Han, J. Zhang, D. Liu, G. Ma, C. Li, L. Liu and D. Kong, ACS Appl. Mater. Interfaces, 2018, 10, 30983–30993 CrossRef CAS.
- K. Heeg and S. Zimmermann, Int. Arch. Allergy Immunol., 2000, 121, 87–97 CrossRef CAS.
- S. Romagnani, Ann. Allergy, Asthma, Immunol., 2000, 85, 9–18 CrossRef CAS.
- M. Saraiva and A. O'Garra, Nat. Rev. Immunol., 2010, 10, 170–181 CrossRef CAS.
- M. Oft, Cancer Immunol. Res., 2014, 2, 194–199 CrossRef CAS.
- A. Naing, J. R. Infante, K. P. Papadopoulos, I. H. Chan, C. Shen, N. P. Ratti, B. Rojo, K. A. Autio, D. J. Wong, M. R. Patel, P. A. Ott, G. S. Falchook, S. Pant, A. Hung, K. L. Pekarek, V. Wu, M. Adamow, S. McCauley, J. B. Mumm, P. Wong, P. Van Vlasselaer, J. Leveque, N. M. Tannir and M. Oft, Cancer Cell, 2018, 34, 775–791 CrossRef CAS.
- H.-M. Wu, J. Wang, B. Zhang, L. Fang, K. Xu and R.-Y. Liu, Life Sci., 2016, 161, 51–59 CrossRef CAS.
- S. M. Kaech and W. Cui, Nat. Rev. Immunol., 2012, 12, 749–761 CrossRef CAS.
- F. Sallusto, J. Geginat and A. Lanzavecchia, Annu. Rev. Immunol., 2004, 22, 745–763 CrossRef CAS.
- L. Liu, P. Ma, H. Wang, C. Zhang, H. Sun, C. Wang, C. Song, X. Leng, D. Kong and G. Ma, J. Controlled Release, 2016, 225, 230–239 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01333a |
| This journal is © The Royal Society of Chemistry 2021 |