DOI:
10.1039/D0BM01328E
(Paper)
Biomater. Sci., 2021,
9, 124-132
Green facile synthesis to develop nanoscale coordination polymers as lysosome-targetable luminescent bioprobes†
Received
9th August 2020
, Accepted 26th September 2020
First published on 29th September 2020
Abstract
Three new coordination polymers (CPs), namely [{M(HL)(L)(H2O)}(ClO4)(H2O)]∞ (M = Zn for CP 1, Mn for CP 2, Cu for CP 3) were synthesized to explore their efficacy as lysosome-targetable luminescent bioprobes. The synthesized CPs were characterized by techniques including single-crystal X-ray analysis, FTIR spectroscopy and elemental analysis. Single-crystal analysis revealed the formation of iso-structural CPs displaying distorted adamantoid topology developed by bridging ligands and H-bonds connections and metals at the nodes. A green hand-grinding technique with a mortar and pestle resulted in nanoscale coordination polymers (NCPs) suitable for cell permeability and was further confirmed by SEM and DLS analyses. Two of these hand-ground nanoscale coordination polymers NCP 1 and NCP 2 showed excellent green luminescence and were explored as potential and selective long-time biotrackers towards lysosome using the human lung carcinoma cell line (A549). Strikingly, the developed bioprobe displayed excellent bio-availability, photostability and excellent selectivity towards lysosomes sustained by various in vitro cell imaging experiments. Moreover, the long-term probing ability of these NCPs turned out to be better than the commercially available lysosome tracker i.e. LysoTracker Red, indicating their potential real-life application in bio-imaging. To the best ofour knowledge, this is the first example of nonexpensive and less toxic essential transition metal-based nanoscale coordination polymers that can behave as effective lysosome-targetable luminescent bioprobes.
Introduction
Coordination polymers (CPs)1–8 are crystalline supramolecular materials formed by an extended array (whether 1D, 2D or 3D) of metal centres linked via multi-topic organic ligands. Research activities on CPs have attracted intriguing attention owing to their potential applications in various fields, such as catalysis,9–11 sensing12–14 metallogelation,15–17 drug delivery,18–24 gas adsorption, separation,25–27 anion separation28,29 and magnetism.30,31 Of late, coordination polymers (CPs) have been extensively used in various biomedical applications because of their versatile properties, such as (a) synthetic amenability, (b) tuneable physicochemical behaviour due to infinite compositional and structural diversities of the used metals as well as ligands and (c) expected ease of excretion by the fast biodegradation of relatively labile covalent coordination bonds. However, most CPs exhibit poor water solubility, and so cannot be effectively administrated in cells. Therefore, the particle size of CPs must be reduced to the nanoregime; usually, the particle size should be below 200 nm.32–37 These nano-CPs (NCPs) could further form colloidal suspensions in suitable biological solvents (in most cases DMSO or PBS buffer) and thus can be easily administrated in cells. Typically, NCPs can be achieved by several techniques, such as sonochemical,38 surfactant-templated,39 nanoscale precipitation,40 solvothermal41 and reverse microemulsion42 syntheses. However, there are very few reports where these NCPs were achieved by a simple mechanical grinding,43–45 which turns out to be a time-economic as well as green synthesis process that avoids the usage of hazardous solvents commonly used in the typical synthesis.46 In recent years, some systematic efforts have been made towards the development of organelle-specific trackers, mainly lysosomal trackers, to visualize molecular recognition in human cells.47,48 In this context, it should be worth mentioning that lysosome plays a key role in various physiological activities, such as cell membrane recycling, metabolism, protein degradation and cell apoptosis.49,50 Various luminescent dyes, importantly rhodamine51 and fluorine,52 have been exclusively used as lysosome trackers as well as imaging agents; although in such conventional tracker probes, one has to worry about the cytotoxicity, poor photostability, poor selectivity and poor solubility of these organic dyes.53 Furthermore, most commercial LysoTrackers, i.e. DND-189, Neutral Red (NR) and DND-99, show high pH dependence, which means they cannot be used significantly as long-term trackers. Indeed, various metal complexes derived mainly from expensive metals, i.e. Au, Ir or Pt, are used as biotrackers for lysosome54–57 However, their poor water solubility as well as poor bioavailability, are the major concerns associated with these metal complexes, and makes them unsuitable for expected biomedical applications. Therefore, developing lower transition metal-based CPs that can behave as efficient long-time lysosome trackers is indeed challenging. With all these challenges in mind, we considered synthesizing various CPs capable of displaying a long-term lysosome tracking property. For this purpose, we chose a pyridyl ligand [4-[(pyridin-2-ylmethylene)-amino]-benzoic acid] (HL) that is capable of being coordinated by both N and O centres so that it could easily form an extended framework by tuning some simple synthetic manipulations. Moreover, various non-expensive transition metal centres, i.e. Zn, Mn and Cu, have been utilized as metal centres because of (a) their superior bio-availability, (b) properties to regulate the metabolic activities of many enzymes and (c) their economic viability. The synthesized CPs were characterized by single-crystal X-ray diffraction (SXRD), elemental analysis, powder X-ray diffraction (PXRD), FTIR and various physico-chemical techniques. To assess the biological activities, the particle-sizes of the CPs were further downsized to the nanoregime by a time-economic facile green synthesis followed by a simple hand-grinding technique. Two of the NCPs turned out to have excellent photoluminescence in the solution state, making them suitable for cellular imaging. Furthermore, the suitability of all these nanoscale CPs as lysosome-targetable luminescent bioprobes was assessed by various in vitro cellular imaging experiments, e.g. co-localization or photobleaching experiments, under biologically optimum conditions. Therefore, we developed a facile green technique to synthesize hand-ground nanoscale coordination polymers displaying selective properties as well as the capability to act as a long-term lysosome tracker.
Experimental section
Materials
All the required chemicals (metal salts and ligands) were purchased from Sigma-Aldrich and used without further purification. The solvents were of HPLC grade and used without any further distillation. The stretching and vibrating frequencies of the as-synthesized and nanoscale CPs were examined by the FTIR (Fourier transform infrared) spectroscopy technique on a PerkinElmer Spectrum Version 10.5.1 using the KBr pellet method and taking air as a background. Elemental analyses (C, H and N) were performed using a PerkinElmer 240C analyzer. The photoluminescence spectra of the dispersed CPs were obtained out on an Hitachi F-7100 fluorescence spectrometer. Scanning electron microscopy (SEM) of the NCPs was carried out with a JEOL JSM-6700F field-emission microscope. DLS studies of the NCPs were carried out in a DLS Malvern particle size analyzer (model no. ZEN 3690 ZETASIZER NANO ZS 90). A549 (Human lung adenocarcinoma) cell lines were purchased from the National Centre for Cell Science, Pune, India. For the cell culture, Dulbecco's modified Eagles’ medium (DMEM), fetal bovine serum (FBS) and antibiotic (1% penicillin/streptomycin) were purchased from Sigma-Aldrich (Merck, Germany), and MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide) and all the other chemicals and reagents used in the cellular studies were purchased from Merck (Germany). A confocal laser scanning microscopy system (Olympus IX 81) was used for the high-quality multi-wavelength fluorescence cellular imaging to study the co-localization and fluorescence recovery after photobleaching.
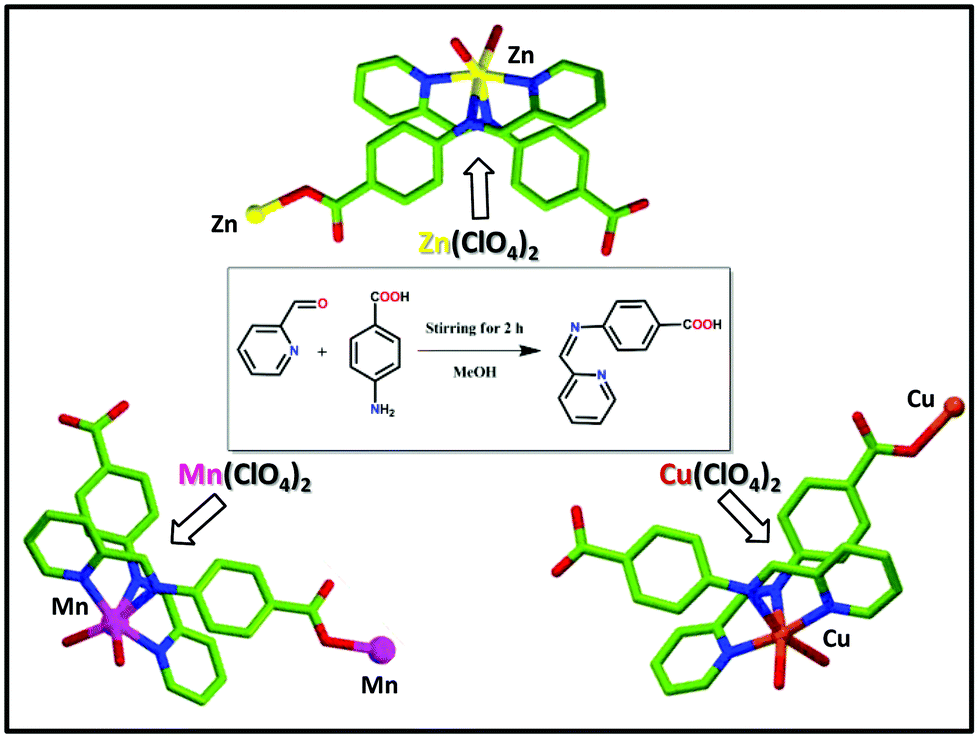
Synthesis of the ligand [4-[(pyridin-2-ylmethylene)-amino]-benzoic acid] (HL)
The ligand (HL) was prepared (Scheme 1) by using 2-formylpyridine and para-aminobenzoic acid (PABA) as the starting reagents using the published method.58 To a methanolic solution of para-aminobenzoic acid (PABA, 1 mmol, 137 mg) 2-formylpyridine (1 mmol, 107 mg) was added drop-wise under stirring for 2 h. After that, the solution was filtered off and washed several times with ether and dried. Yield: 80%. 1H NMR (300 MHz, DMSO-d6) (Fig. S1, in the ESI†) δ 12.53 (1H, brs), 8.61–8.84 (1H, m), 8.17–8.20 (1H, m), 7.84–8.01 (2H, m), 7.74–7.68 (1H, m), 7.59–7.65 (1H, m), 7.55–7.58 (1H, m), 7.39–7.41 (1H, m), 7.23–7.39 (1H, m); 13C NMR (75 MHz, DMSO-d6) (Fig. S2, in ESI†) δ 194.2, 167.8, 163.0, 154.9, 150.2, 137.6, 131.7, 131.4, 128.8, 122.1. ESI-MS (ES+, methanol) calcd (Fig. S3, in ESI†) m/z for C13H11N2O2+: 226.23 amu; found 227.4728 amu. FTIR (4000–400 cm−1) (Fig. S4, in ESI†): 3447 (w), 3045 (w), 2785 (w), 2600 (w), 2492 (w), 1906 (w), 1700 (s), 1602 (s), 1483 (s), 1418 (s), 1226 (w), 1157 (s), 1070 (s), 995 (s), 853 (s), 789 (s), 669 (s), 528 (s), 452 (w).
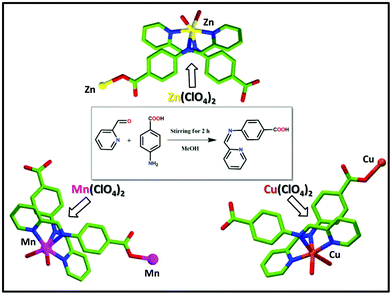 |
| | Scheme 1 Schematic representation including the preparation ligand (L) and coordination polymer 1 (CP 1), coordination polymer 2 (CP 2) and coordination polymer 3 (CP 3). | |
Synthesis of the coordination polymer 1 (CP 1)
The ligand (HL) (0.5 mmol, 113 mg) was dissolved in a mixture of 10 mL MeOH and 5 mL EtOH mixture. A 2 mL aqueous solution of Zn(ClO4)2 (0.5 mmol, 186 mg) was added to it drop-wise with stirring. Then the resultant mixture was refluxed for 3 h. Upon slow cooling, a room temperature yellowish solution was filtered into a beaker and kept at room temperature. After 7 days yellow block-shaped crystals were obtained (yield 87% based on L), which were then filtered, washed with ether several times, and dried under air. FTIR (4000–400 cm−1) (Fig. S5, in the ESI†): 3400 (w), 3098 (w), 2928 (w), 2600 (w), 2500 (w), 2015 (w), 1676 (s), 1598 (s), 1530 (s), 1395 (s), 1259 (s), 1079 (s), 920 (s), 864 (s), 774 (s), 695 (w), 605 (s), 514 (s), 413 (w). Elemental analysis: C26H23ClZnN4O10 (652.30): calcd C 47.87, H 3.55, N 8.59; found C 47.78, H 3.32, N 8.51.
Synthesis of coordination polymer 2 (CP 2)
Coordination polymer 2 (CP 2) was synthesized following a similar method as described for CP 1 by using Mn(ClO4)2 (0.5 mmol, 181 mg) as a metal salt instead of Zn(ClO4)2 salt. Brown block-shaped crystals were obtained (yield 83% based on L), which were further washed with ether and dried in air. FTIR (4000–400 cm−1) (Fig. S6, in the ESI†): 3358 (vw), 3064 (w), 2939 (w), 2624 (w), 2500 (w), 1992 (w), 1687 (s), 1575 (s), 1507 (s), 1372 (s), 1259 (s), 1168 (s), 1090 (w), 910 (s), 876 (s), 774 (w), 672 (w), 616 (s), 526 (s), 425 (w). Elemental analysis: C26H23ClMnN4O10 (641.87): calcd C 48.65, H 3.61, N 8.73; found C 48.48, H 3.73, N 24.82.
Synthesis of coordination polymer 3 (CP 3)
Coordination polymer 3 (CP 3) was synthesized following a similar method as that was performed for CP 1. Here, only instead of Zn(ClO4)2, Cu(ClO4)2 (0.5 mmol, 131 mg) was used as a metal salt. Green block-shaped crystals were obtained (yield 85.5% based on L), which were further washed with ether and dried in air. FTIR (4000–400 cm−1) (Fig. S7, in the ESI†): 3436 (vw), 2454 (w), 1992 (w), 1913 (w), 1699 (s), 1609 (s), 1507 (s), 1409 (w), 1293 (s), 1225 (s), 1158 (s), 1090 (s), 1022 (s), 920 (s), 853 (s), 751 (w), 662 (w), 526 (w), 458 (w). Elemental analysis: C26 H23ClCuN4O10 (650.48): calcd C 48.01, H 3.56, N 8.61; found C 47.97, H 3.63, N 8.56.
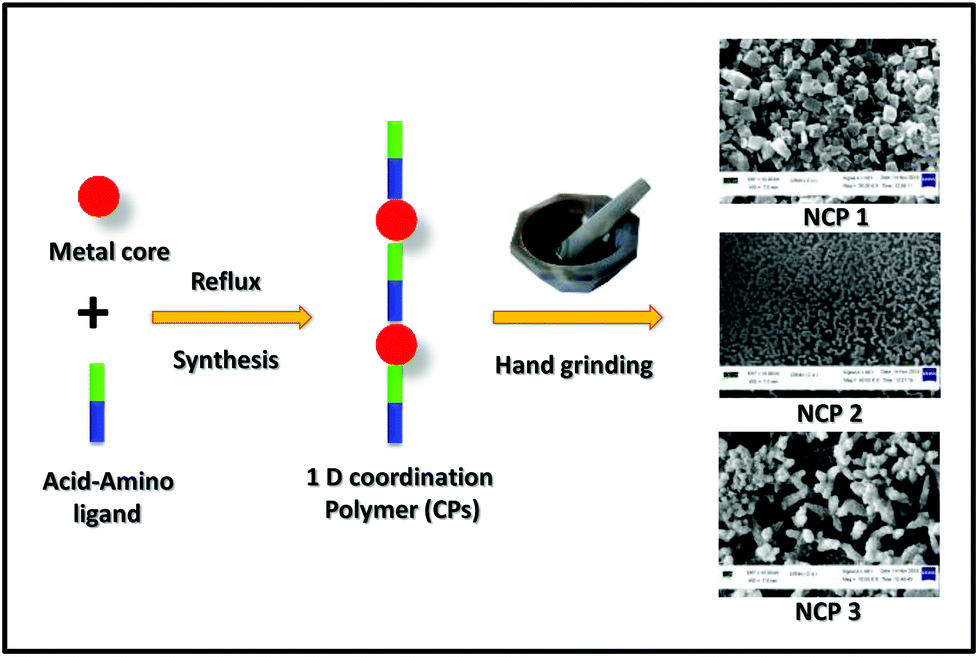
Synthesis of nanocoordination polymers (NCPs)
A small portion (nearly 50–60 mg) of the synthesized CPs (1–3) was taken separately in a mortar. By using a pestle, each portion was being ground manually for 20 min. Herein, nano-sized CPs were ready to use directly in the several studies that are mentioned here.
Preparation of the SEM sample
All the scaled-down CPs (1–3) were dispersed in water through sonication for about 45 min. The dispersed solutions were drop-cast on separate glass disks. The disks were then dried under air in an incubator at 60 °C for 1 day, and subjected to an SEM experiment to record the corresponding images.
Preparation of the sample for the powder X-ray diffraction analysis
The finely powdered samples (∼30 mg) were smeared over a glass slide. Powder X-ray experiments were performed on a powder X-ray diffractometer (Model: X'Pert Powder, PANalytical) using Cu–K Alpha radiation in theta-2theta geometry in the 5°–35° 2θ range using a 0.02° step size per second.
Preparation of the sample for the DLS experiment
1 mg of each nanoscale CP was dispersed in 4 mL water. The dispersed solutions were taken in a quartz cuvette and to get the radius distribution of the solvent-coated NCPs dynamic light scattering (DLS) experiments were performed.
Cell cultures
A549 cells were cultured in Dulbecco's modified eagle's medium (DMEM) supplemented with penicillin (100 U mL−1), streptomycin (100 μg mL−1) and L-glutamine (2.5 × 10−4 M) in a humidified atmosphere.
MTT assay
The cytotoxicity of the NCPs was evaluated by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay following a published protocol.59 In a typical experiment, 0.5 × 104 cells per mL were seeded in 96-well plates and incubated for 24 h at 37 °C with 5% CO2. After 24 h, the cell medium was sucked out and the cells were charged with different concentrations of (2.5, 5, 10, 20 and 40 μM) of NCP 1, 2 and 3 and further incubated for 24 h. Next, the cells were incubated with MTT (0.5 mg mL−1) for 3 h in a humidified condition. The purple-coloured crystals, produced by the reaction of mitochondrial reductase of living cells with MTT, were then dissolved in 100 μL DMSO. The cells were then kept for 45 min. The absorbance of the purple-coloured solution, indicating the number of living cells, was assessed using a microplate reader at 570 nm.
Luminescence imaging
A459 cells were washed with PBS thoroughly and charged with 10 μM of NCPs in culture medium (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :99 of DMSO:culture media). The cells were kept in humidified conditions for 6 h. Next, the cells were again washed with PBS solution. The cells then produced green fluorescence centred at 500–530 nm when excited at 458 nm with a semiconductor laser. Overlap images were also obtained between the LysoTracker Red and the NCPs by the corresponding luminescent intensities within the selected regions of interest. To perform the colocalization experiments, the cells were treated with 10 μM of NCPs and incubated for 6 h. LysoTracker Red was added just before cell imaging in the same concentration.55 The cells were excited at 458 nm for the NCPs, whereas the LysoTracker Red-treated cells were excited at 560 nm.
:99 of DMSO:culture media). The cells were kept in humidified conditions for 6 h. Next, the cells were again washed with PBS solution. The cells then produced green fluorescence centred at 500–530 nm when excited at 458 nm with a semiconductor laser. Overlap images were also obtained between the LysoTracker Red and the NCPs by the corresponding luminescent intensities within the selected regions of interest. To perform the colocalization experiments, the cells were treated with 10 μM of NCPs and incubated for 6 h. LysoTracker Red was added just before cell imaging in the same concentration.55 The cells were excited at 458 nm for the NCPs, whereas the LysoTracker Red-treated cells were excited at 560 nm.
Single-crystal X-ray crystallography
Single crystals of all these CPs were obtained by a solvothermal process. Data were recorded using MoKα (λ = 0.71073 Å) radiation on a BRUKER diffractometer equipped with a CCD area detector. Data collection and reduction of the data sets were carried out using software in the Bruker Smart Apex and Bruker Saint packages.60 All the structures of the CPs were solved by direct methods61 and refined with the SHELX program62 with the contribution of H atoms fixed at the geometrical positions. All the non-hydrogen atoms were treated anisotropically. In CP 1 and Cp 3, the oxygen atoms of ClO4 anions were found to be disordered over two positions and refined with occupancy factors of 0.586(19)/0.414(19) and 0.513(17)/0.487(17), respectively. The CCDC no. are 2006521, 2006522 and 2006523† for CP 1, CP 2 and CP 3, respectively.
Results and discussion
Crystal structure description
The solvothermal reaction of the ligand HL with MCl2 (M = Zn, Mn, Cu) yield three new isostructural coordination polymers (CPs) having the basic structural formula [{M(HL)(L)(H2O)}(ClO4)(H2O)]∞ (M = Zn for 1, Mn for 2, Cu for 3). The single-crystal XRD analyses reveal that all the compounds crystallized in the orthorhombic Pbca space group with comparable unit cell dimensions (Table S1, in the ESI†).
Structural description
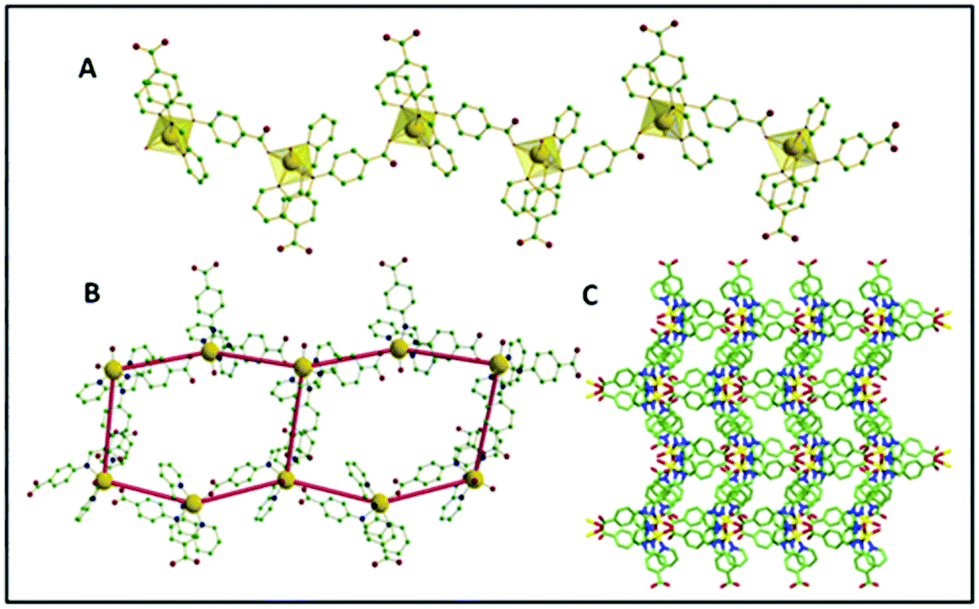
The asymmetric units were constituted by one central M(II) ion (M = Zn, Mn, Cu for 1, 2 and 3, respectively), two HL ligands (of which one was deprotonated), two water molecules (one coordinated and one lattice occluded) and one non-coordinated perchlorate ion. The M–O/N bond distances were 2.038(3)–2.267(3) Å (for Zn), 2.088(2)–2.339(2) Å (for Mn complex) and 2.052(10)–2.179 (13) Å (for Cu complex), where the longest values refer to the imino nitrogen atoms. In every case, the M centres exhibited a distorted octahedral geometry, where four positions of the octahedron were chelated by the pyridyl and imine nitrogen atoms from the two ligands (HL, L-), and the other two positions were coordinated with one carboxylate oxygen of a symmetry-related deprotonated L- ligand and one water molecule. Considering the isostructural feature of the CPs, the detailed structural elucidation of CP 1 has been further demonstrated herein. Interestingly it was observed that the ligand HL majorly plays two crucial roles during crystallization: (i) it acts as an N-donor chelating the ligand to saturate the secondary valency/valence of the central metal ion, (ii) it participates as an N,O-donor divergent linker to construct the polymeric network. In the crystal structure, one of the HL ligands undergoes deprotonation to form extended coordination with the adjacent Zn centre to produce an infinite one-dimensional (1D) helical framework. Each helical chain further propagates two-dimensionally forming a hexagonal architecture through various hydrogen bonding interactions sustained by the water molecules (both coordinated and lattice occluded) and carboxylate oxygen atoms. Such a special arrangement is further stabilized via π⋯π (centroid to centroid distance 3.837 Å) and CH⋯π (CH to centroid distance 3.309 Å) interactions between contagious phenyl and pyridyl rings of the acid molecules in the native skeleton (Fig. 1).
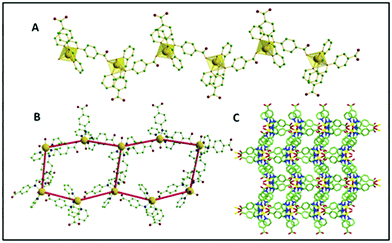 |
| | Fig. 1 (A) 1D polymeric chain of CP 1 with the polyhedral view around the central Zn; (B) the hexagonal unit formation through hydrogen bonding; (C) crystal packing along the crystallographic ‘a’ axis. | |
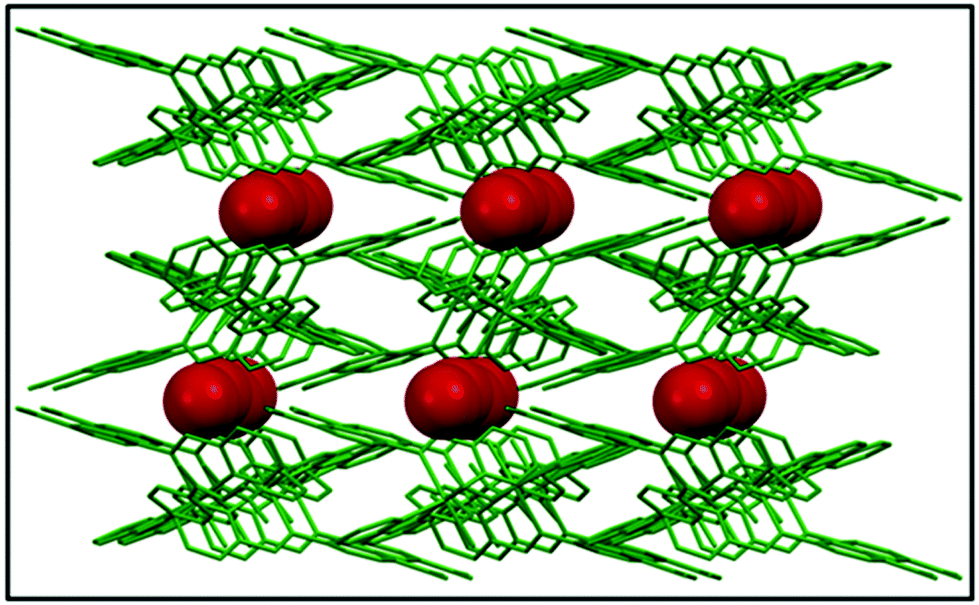
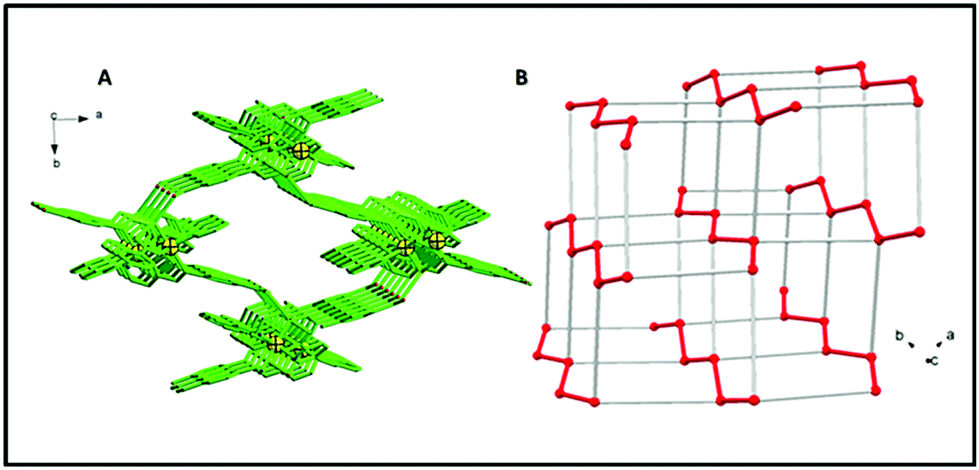
A continuous chain-like arrangement of lattice water molecules could be observed throughout the structure, which threaded the simultaneous 1D layer as mentioned above (Fig. 2). The water molecule O1w forms an H-bond (O2w⋯O2 of 2.62 Å) that reinforces the structure and besides is involved in a second H-bond with O4 (O1w⋯O4′ of 2.80 Å) to form a 3D architecture (Fig. 3A). The 3D network presents a distorted adamantoid topology formed by bridging ligands and H-bonds connections and metals at the nodes (Fig. 3B). Similar structural features were witnessed in the other two CPs (2 and 3). The immediate coordination environments of Mn and Cu in CP 2 and 3 with another required bond angle, bond length and relevant H-bonding parameters are summarized in Fig. S8–S15 and Tables S2–S8 in the ESI.†
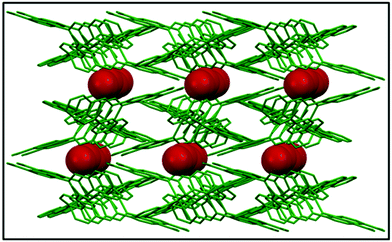 |
| | Fig. 2 The infinite water chain within the crystal structure viewed along the crystallographic ‘b’ axis. | |
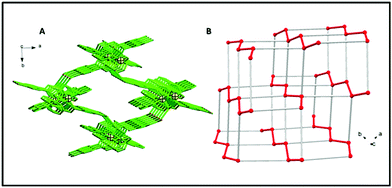 |
| | Fig. 3 (A) 3D structure viewed down axis c and built by H bonds connecting the polymeric coordination chains. (H atoms, lattice water and perchlorate anions are not shown for picture clarity.) (B) Representation of the distorted demantoid topology with metals at the nodes. Red lines indicate the coordination polymers (Zn–Zn distance = 9.585(3) Å), grey lines are connections built by H-bonds (Zn–Zn distance = 10.815(3) Å). | |
Preparation of the nanoscale coordination polymers (NCPs)
Since our main aim was to develop transition metal-based CPs displaying a capability as efficient and selective long-time Lysosome trackers; at this moment it was of the utmost important to downscale the CPs into the nanoregime. Therefore, those NCPs (Scheme 2) could have increased solubility in commonly used biomedical solvents, i.e. DMSO, PBS buffer solvent. Interestingly, grinding by a mortar and pestle for 20 min resulted in nanoscale coordination polymers (NCPs).
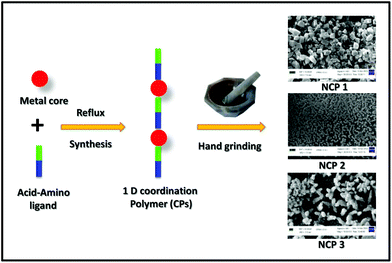 |
| | Scheme 2 Schematic representation for the preparation of the nano coordination polymers using a hand-grinding technique. | |
All these NCPs were then characterized by SEM images by drop-casting a water-dispersed solution of the NCPs on glass disks; nanoscale morphologies were obtained for NCP 1 (∼100 nm) and NCP 2 (∼180 nm), whereas for NCP 3, an aggregated structure was observed. A close inspection of the SEM images showed that NCP 1 acquired a uniformly distributed hexagonal shape (Fig. 4A) throughout the region, with a ∼100 nm particle size, whereas NCP 2 adopted a round-network-like morphology (Fig. 4B), which was furthered made up of small rod-shaped particles having widths of ∼150–180 nm. However, NCP 3 exhibited an aggregated morphology (Fig. 4C), having a particle size of ∼0.8 μm. Dynamic light scattering (DLS) data further confirmed the formation of NCPs; the hydrodynamic radii were found to be 162, 207 and 826 nm for NCP 1, NCP 2 and NCP 3, respectively (Fig. S16, in the ESI†). Therefore, the particle sizes for NCP 1 (∼100 nm) and NCP 2 (∼180 nm) were in the nanoregime; which could further increase their suitability as biotrackers towards Lysosomes. To check the structural integrity of all these NCPs, various physico-chemical experiments, i.e. FTIR spectroscopy, PXRD, EDX analysis, were employed. The chemical structures as well as the metal–ligand interactions of all three-nanoscale coordination polymers remained unaltered, as indicated by FTIR spectroscopy (Fig. S5–S7, in the ESI†). Powder X-ray diffraction (PXRD) further indicated the crystalline integrity of the NCPs. The crystalline phase purity all three nanoscale coordination polymers were found to be excellent since the PXRD diffraction pattern of the NCPs exactly matched with the simulated as well as the bulk materials of the corresponding CPs (Fig. S17–S29, in the ESI†). The chemical composition of NCPs was demonstrated by energy dispersive X-ray (EDX) spectrometry studies (Fig. S20–S22, in the ESI†). The EDX analysis confirmed that NCP 1 consisted of C (95.37%, atomic %), N (0.90%, atomic %), O (3.26%, atomic %), Cl (0.45%, atomic %) and Zn (0.02%, atomic %). Both NCP 2 and NCP 3 contained the same elements but having different metals: Mn (in the case of NCP 2, having 0.71%, atomic %) and Zn (in the case of NCP 3, having 10.86%, atomic %). UV-Vis spectroscopy of these NCPs was performed to obtain insights into their organization in solution (Fig. S23†).
 |
| | Fig. 4 SEM images of NCPs: (A) NCP 1 adopted a hexagonal shape, (B) NCP 2 had a rod-shaped morphology and (C) NCP 3 adopted an aggregated clumsy morphology. | |
Photoluminescence studies of the nanoscale coordination polymers
Owing to converting the corresponding NCPs, the next step was to check the suitability of these NCPs as bioprobes. For this purpose, the photophysical properties of the NCPs were assessed. In a typical experiment, DMSO/PBS buffer (1/99 v/v; pH 7) suspensions of the NCPs displayed significant emission centred at ∼500–530 nm (Fig. 5). Interestingly, NCP 1 (λem ∼ 530 nm) and NCP 2 (λem ∼ 500 nm) produced intense photoluminescence in the green region when excited with blue light (λex ∼ 480 nm); thereby both NCP 1 as well as NCP 2 could be further utilized as promising candidates for live-cell imaging.
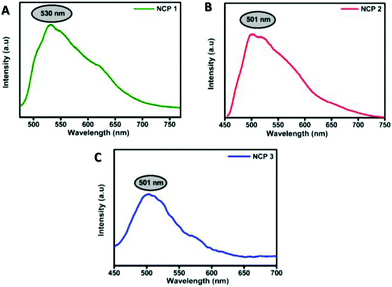 |
| | Fig. 5 Photoluminescence spectra of the NCPs (1–3) in solution phase dispersed in PBS buffer solution. | |
NCP 1 displayed significantly higher fluorescence intensity compared to the others, as attributed by the quantum yield calculations depicted in Fig. S24 and Table S9 in the ESI.† The quantum yield of NCP 1 (0.015) was found to be much higher compared to NCP 2 (0.008) followed by NCP 1 (0.003); thereby making both NCP 1 and 2 more suitable for further cell-imaging application. The detection limit for NCP 1 was 0.325 μM (Fig. S25 and Table S10†), which is very much promising considering the applied in vitro dose concentration.63
Biological studies
The foregoing discussions revealed the successful development of NCPs based on transitional metals. Since these NCPs displayed significant emission intensity in the green region, our next step was to check the suitability of all these NCPs as efficient long-term biotrackers towards lysosome. For this purpose, A549 cell lines were chosen, as these cancer cells tend to show enhancements in lysosomal volume and expression.64–66
Cytotoxicity assessment of the NCPs
Before performing cellular imaging, cytotoxicity profiling was performed to understand the lethality, and to assess the viable doses of all the three NCPs.67 The cytotoxicity profiles of all three NCPs were obtained in A549 cells using the MTT assay following a published protocol. The MTT assay revealed that all the NCPs studied had a very low interference with cellular toxicity. NCP 1 displayed ∼100% cell viability whereas ∼90% cell viability was observed for NCP 2 when the cells were treated with a concentration range from 0 to 50 μm. Herein, it is worth mentioning that the cell viability of non-treated A549 cells was considered to be 100% (Fig. 6). The MTT assay data revealed that further cellular imaging could be done in between the concentration ranging from 0 to 50 μM.
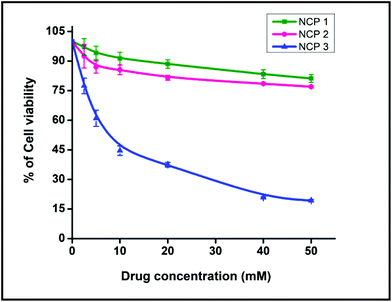 |
| | Fig. 6 Cell viability was measured with the MTT assay. Data represent the means of three independent experiments. | |
Intracellular luminescence study
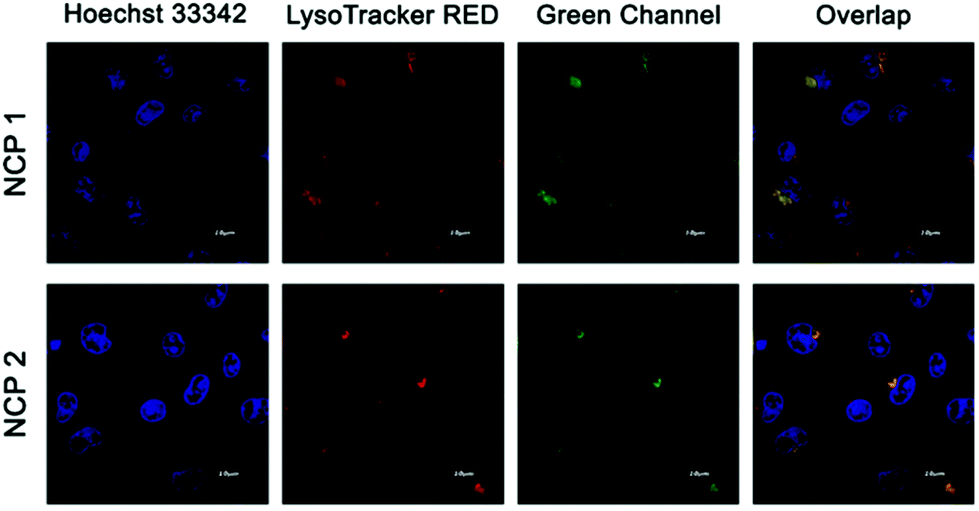
Since all three NCPs displayed significant emission in the green channel, the NCPs were next subjected to confocal luminescence imaging using A549 cells.68 In a typical experiment, intense green intracellular luminescence centred at 500–530 nm was observed in lysosomes under a confocal microscope when the cells were treated with 10 μM of NCPs (Fig. 7 for NCP 1 and NCP 2, Fig. S26, in the ESI† for NCP 3). To ensure the labelling specificity of all these NCPs, co-localization experiments were performed by co-staining the A549 cells with both LysoTracker Red and NCP 1 or 2. The extensive overlapping with Lysotracker was observed for both NCP 1 and 2.69 Luminescence imaging further demonstrated that NCP 1 and NCP 2 were nearly co-localized with the LysoTracker in lysosomes, whereas NCP 3 displayed no such significant co-localization. The specific localization of the depicted NCPs indicated that both the compounds may be further used as a probe for imaging lysosome. Since NCP 1 and 2 could serve as a potential probe for lysosome, further biological experiments were carried out with NCP 1 and 2.
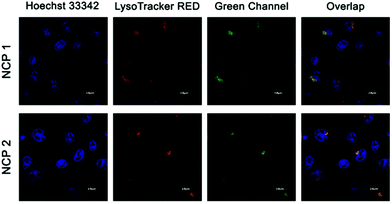 |
| | Fig. 7 Confocal images of A549 cells incubated with NCP 1 and 2 and LysoTracker Red indicating colocalization. Red channel emission was collected in the range of 580–620 nm, whereas the green channel emission was collected in the range of 500–530 nm. | |
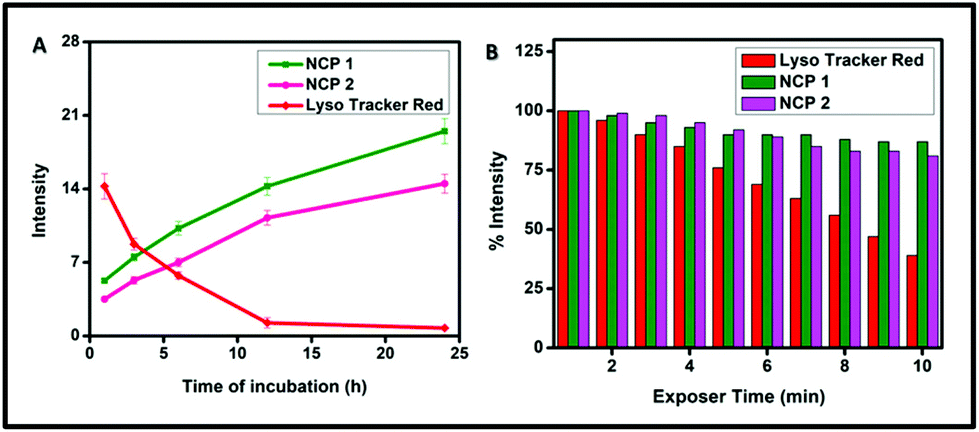
To ensure the suitability of NCPs as long-term lysosome trackers, A549 cells were further treated with NCP 1 and 2 following a pre-suggested study, and cellular images were captured at different time intervals up to 24 h.70 The results revealed that the luminescence intensity of the cells increased with increasing the incubation time, whereas the intensity decreased gradually for LysoTracker Red (Fig. 8). The images further demonstrated that the maximum luminescence intensity was observed after ∼12 h of incubation. Interestingly, the cells retained the maximum fluorescence intensity for up to 24 h of incubation. Such increasing fluorescence intensity may be attributed to the continuous cellular uptake nature of NCP 1 and 2 in the A459 cell line. However, for the LysoTracker Red-treated cells, there was a gradual decrease in fluorescence intensity, as illustrated by the quantification of the fluorescence intensity experiment (Fig. 9A) with time. Therefore, it can be concluded that both the NCPs could be used as potential long-term lysosome trackers.
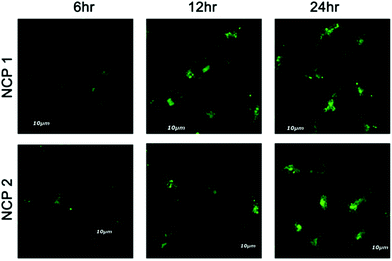 |
| | Fig. 8 Time-dependent luminescence intensity study for NCP 1 and NCP 2 incubated with A549 cells for different time intervals. | |
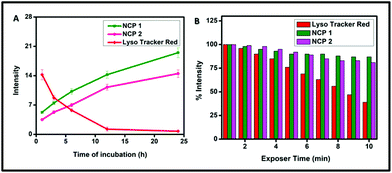 |
| | Fig. 9 (A) Luminescence intensity quantification for LysoTracker Red, NCP 1 and NCP 2 incubated with A549 cells for time intervals of 0 to 24 h, (B) percentage luminescence intensity loss for LysoTracker Red, NCP 1 and NCP 2 in the A549 cell line to study the compounds’ photobleaching with increasing time. | |
Next, photobleaching experiments were performed according to a study reported by Tran et al. for both the NCPs (1 and 2) to ensure their photostability.71 The results are depicted in Fig. 9B, which demonstrated that there was no significant loss of fluorescence intensity for both the NCP 1- and 2-treated cells, whereas, for LysoTracker Red, the intensity gradually decreased. The percentage luminescence intensity was measured considering the brightest intensity signal as 100%. The results revealed that NCP 1 and NCP 2 retained more than 80% of the luminescence signals with continuous excitation for 10 min. However, LysoTracker Red lost almost 45% of its fluorescence intensity. Therefore, both the NCPs exhibited better photostability than the commercially available bioprobe LysoTracker Red.
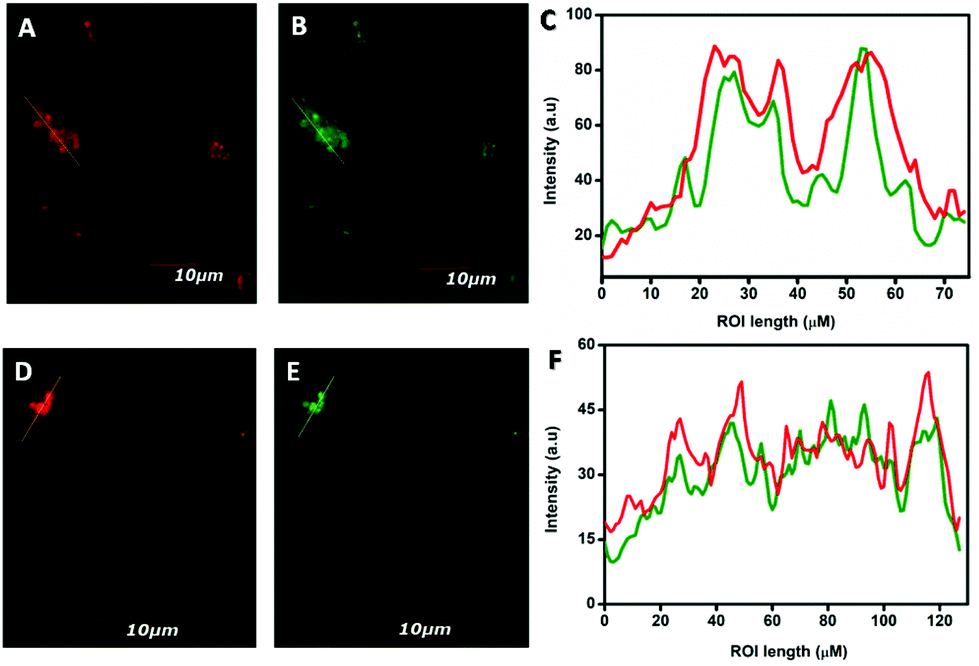
Finally, the fluorescence intensities of the ROIs across the cells were examined by incubating A549 cells with 10 μM of both NCP 1 and 2 in humidified conditions for 6 h. The intracellular fluorescence was observed by using colocalization experiments followed by co-incubating A549 cells with both NCP 1 or 2 and LysoTracker Red, as depicted previously (vide supra). The results revealed that the green fluorescence of both the NCPs overlapped nearly with the red fluorescence of LysoTracker Red (Fig. 10A, B, and D, E). Moreover, the fluorescence intensities of the ROIs across the cells with red channel and green channel emission were collected using the ImageJ software and compared through histograms (Fig. 10C and F).
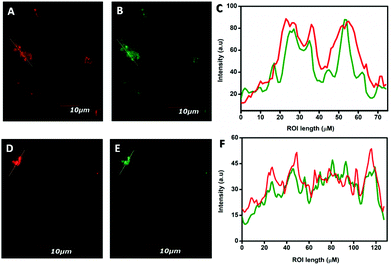 |
| | Fig. 10 Fluorescence intensities across the regions of interest (ROIs) over the cells incubated with LysoTracker Red, NCP 1 and 2, respectively, were measured (A, B, D, and E) and compared (C and F). | |
Biostability tests of all the NCPs were performed in PBS buffer incubated for 24 h in humidified conditions. The SEM images and DLS study indicated that the NCPs sustained their structural integrity (Fig. S27, S28 and S29 in the ESI†).
The structural integrity of the synthesized NCP 1 was also confirmed via PXRD (Fig. S30†) and time-dependent DLS studies in lysosomal pH (Fig. S31†).
The foregoing discussions demonstrated the successful construction of nano-scaled CPs, displaying excellent green luminescence, which were employed as selective bioprobes towards lysosomes followed by in vitro cell imaging experiments. The cell imaging data further revealed that NCP 1 and 2 were more effective than the corresponding NCP 3, which corroborated well with their corresponding particle sizes (particle size of NCP 1 ∼ 100 nm, NCP 2 ∼ 150–180 nm and NCP 3 ∼ 0.8 μm, respectively) and also their fluorescence intensity as sustained by photoluminescence data.
Conclusions
In summary, we developed a facile green hand-grinding technique towards the design of three nanoscale crystalline materials that could serve as selective biotrackers for intracellular lysosome. For this purpose, three new CPs were synthesized and characterized by various physico-chemical experiments, including FTIR and single-crystal XRD. Herein, we deliberately chose no expensive and less toxic essential transitional metals, i.e. ZnII, MnII and CuII, as the metal centres. To afford biomedical suitable nanoregime particles, a time-economic and green, facile hand-grinding synthesis was employed; the NCPs retained their structural as well as compositional integrity. NCP 1 and 2 displayed significant photoluminescence in the green region when excited with blue light; thereby making them promising candidates for bioimaging. Interestingly, the synthesized NCPs displayed significant bio-availability in A459 cells over the concentration range from 0 to 50 μM. Moreover, the NCPs displayed specific-localization towards lysosome and provided better photostability compared to the commercially available bioprobe Lysosome Tacker Red; thereby these luminescent NCPs could be employed as organ-specific long-term probes of lysosomes. These unique NCPs open up a new avenue for the development of selective long-term organ-specific live-cell imaging agents.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. K. would like to acknowledge DST-INSPIRE Fellowship [(No. DST/INSPIRE Fellowship/2018/IF180018 and Application Reference Number DST/INSPIRE/03/2017/001850)]. S. M. thanks CSIR, New Delhi for the research fellowship (CSIR Grant no.: 09/028(1009)/2017-EMR-I). R. P. thanks CSIR, New Delhi for CSIR-RA fellowship (CSIR grant number: 09/028(1089/2019-EMR-I). The authors would like to thank CRNN and the department of physics, C. U. for providing SEM, TEM, PXRD facilities. We would like to thank Department of Bioscience and Bioengineering, Indian Institute of Technology Bombay for providing the biological facilities and we would also like to thank Nucleus Medical Media, USA for their support.
References
- A. Schoedel, M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116, 12466–12535 CrossRef CAS.
- H. C. J. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- Z. Zhang and M. J. Zaworotko, Chem. Soc. Rev., 2014, 43, 5444–5455 RSC.
- N. N. Adarsh and P. Dastidar, Chem. Soc. Rev., 2012, 41, 3039–3060 RSC.
- D. Mok Shin, I. Su Lee, Y. Keun Chung and M. Soo Lah, Chem. Commun., 2003, 3, 1036–1037 RSC.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- J. K. Schnobrich, O. Lebel, K. A. Cychosz, A. Dailly, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2010, 132, 13941–13948 CrossRef CAS.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS.
- A. Corma, H. García and F. X. Llabrés I Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS.
- M. Rimoldi, A. J. Howarth, M. R. Destefano, L. Lin, S. Goswami, P. Li, J. T. Hupp and O. K. Farha, ACS Catal., 2017, 7, 997–1014 CrossRef CAS.
- I. H. Park, R. Medishetty, J. Y. Kim, S. S. Lee and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 53, 5591–5595 CrossRef CAS.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS.
- K. Jayaramulu, R. P. Narayanan, S. J. George and T. K. Maji, Inorg. Chem., 2012, 51, 10089–10091 CrossRef CAS.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS.
- J. Zhang and C. Y. Su, Coord. Chem. Rev., 2013, 257, 1373–1408 CrossRef CAS.
- S. Samai and K. Biradha, Chem. Mater., 2012, 24, 1165–1173 CrossRef CAS.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS.
- P. Rivera-Fuentes and S. J. Lippard, Acc. Chem. Res., 2015, 48, 2927–2934 CrossRef CAS.
- J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS.
- M. Paul and P. Dastidar, Chem. – Eur. J., 2016, 22, 988–998 CrossRef CAS.
- Z. Ma and B. Moulton, Coord. Chem. Rev., 2011, 255, 1623–1641 CrossRef CAS.
- D. Liu, C. Poon, K. Lu, C. He and W. Lin, Nat. Commun., 2014, 5, 1–11 CrossRef.
- M. H. Teplensky, M. Fantham, P. Li, T. C. Wang, J. P. Mehta, L. J. Young, P. Z. Moghadam, J. T. Hupp, O. K. Farha, C. F. Kaminski and D. Fairen-Jimenez, J. Am. Chem. Soc., 2017, 139, 7522–7532 CrossRef CAS.
- H. Sato, W. Kosaka, R. Matsuda, A. Hori, Y. Hijikata, R. V. Belosludov, S. Sakaki, M. Takata and S. Kitagawa, Science, 2014, 343, 167–170 CrossRef CAS.
- Y. B. Zhang, H. Furukawa, N. Ko, W. Nie, H. J. Park, S. Okajima, K. E. Cordova, H. Deng, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 2641–2650 CrossRef CAS.
- S. I. Noro, S. Kitagawa, M. Kondo and K. Seki, Angew. Chem., Int. Ed., 2000, 39, 2081–2084 CrossRef.
- R. Custelcean and B. A. Moyer, Eur. J. Inorg. Chem., 2007, 2007, 1313–1313 CrossRef.
- R. Custelcean, Chem. Soc. Rev., 2014, 43, 1813 RSC.
- R. Bikas, H. Hosseini-Monfared, V. Vasylyeva, J. Sanchiz, J. Alonso, J. M. Barandiaran and C. Janiak, Dalton Trans., 2014, 43, 11925 RSC.
- W. Zhang and R. G. Xiong, Chem. Rev., 2012, 112(2), 1163–1195 CrossRef CAS.
- K. Lu, C. He, N. Guo, C. Chan, K. Ni, R. R. Weichselbaum and W. Lin, J. Am. Chem. Soc., 2016, 138, 12502–12510 CrossRef CAS.
- C. He, D. Liu and W. Lin, Chem. Rev., 2015, 115(19), 11079–11108 CrossRef CAS.
- Y. Lu, B. Yan and J. L. Liu, Chem. Commun., 2014, 50, 9969 RSC.
- A. Foucault-Collet, K. A. Gogick, K. A. White, S. Villette, A. Pallier, G. Collet, C. Kieda, T. Li, S. J. Geib, N. L. Rosi and S. Petoud, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17199–17204 CrossRef CAS.
- W. Morris, W. E. Briley, E. Auyeung, M. D. Cabezas and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 7261–7264 CrossRef CAS.
- K. Sarkar and P. Dastidar, Chem. – Eur. J., 2016, 22, 1–13 CrossRef.
- E. Haque, N. A. Khan, H. J. Park and S. H. Jhung, Chem. – Eur. J., 2010, 16, 1046–1052 CrossRef CAS.
- K. M. L. Taylor, A. Jin and W. Lin, Angew. Chem., Int. Ed., 2008, 47, 7722–7725 CrossRef CAS.
- W. J. Rieter, K. M. Pott, K. M. L. Taylor and W. Lin, J. Am. Chem. Soc., 2008, 130, 11584–11585 CrossRef CAS.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS.
- W. J. Rieter, K. M. L. Taylor, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2006, 128, 9024–9025 CrossRef CAS.
- T. Kundu, S. Mitra, P. Patra, A. Goswami, D. Díazdíaz and R. Banerjee, Chem. – Eur. J., 2014, 20, 10514–10518 CrossRef CAS.
- M. Paul, K. Sarkar, J. Deb and P. Dastidar, Chem. – Eur. J., 2017, 23, 5736–5747 CrossRef CAS.
- S. Mukherjee, S. Ganguly, K. Manna, S. Mondal, S. Mahapatra and D. Das, Inorg. Chem., 2018, 57(7), 4050–4060 CrossRef CAS.
- H. Yang, C. Qin, C. Yu, Y. Lu, H. Zhang, F. Xue, D. Wu, Z. Zhou and S. Yang, Adv. Funct. Mater., 2014, 24, 1738–1747 CrossRef CAS.
- B. Zhang, C. Ge, J. Yao, Y. Liu, H. Xie and J. Fang, J. Am. Chem. Soc., 2015, 137(2), 757–769 CrossRef CAS.
- M. Fernández-Suárez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929–943 CrossRef.
- G. Kroemer and M. Jäättelä, Nat. Rev. Cancer, 2005, 5, 886–897 CrossRef CAS.
- L. He, C. P. Tan, R. R. Ye, Y. Z. Zhao, Y. H. Liu, Q. Zhao, L. N. Ji and Z. W. Mao, Angew. Chem., 2014, 126, 12333–12337 CrossRef.
- Z. Li, Y. Song, Y. Yang, L. Yang, X. Huang, J. Han and S. Han, Chem. Sci., 2012, 3, 2941–2948 RSC.
- X. Wang, D. M. Nguyen, C. O. Yanez, L. Rodriguez, H. Y. Ahn, M. V. Bondar and K. D. Belfield, J. Am. Chem. Soc., 2010, 132(35), 12237–12239 CrossRef CAS.
- J. Li, N. Kwon, Y. Jeong, S. Lee, G. Kim and J. Yoon, ACS Appl. Mater. Interfaces, 2018, 10(15), 12150–12154 CrossRef CAS.
- D. G. Smith, B. K. Mc Mahon, R. Pal and D. Parker, Chem. Commun., 2012, 48, 8520–8522 RSC.
- C. Y. Liu, X. R. Wei, Y. Chen, H. F. Wang, J. F. Ge, Y. J. Xu, Z. G. Ren, P. Braunstein and J. P. Lang, Inorg. Chem., 2019, 58(6), 3690–3697 CrossRef CAS.
- T. M. George, M. S. Krishna and M. L. P. Reddy, Dalton Trans., 2016, 45, 18719–18729 RSC.
- K. Qiu, H. Zhu, T. W. Rees, L. Ji, Q. Zhang and H. Chao, Coord. Chem. Rev., 2019, 398, 113010 CrossRef.
- D. Xue, Q. X. Peng and S. Z. Zhan, Inorg. Chem. Commun., 2017, 82, 11–15 CrossRef CAS.
- Q. S. Wang, Y. Xiang, Y. L. Cui, K. M. Lin and X. F. Zhang, PLoS One, 2012, 7, e34122 CrossRef CAS.
-
SAINT, SMART, Software Reference Manual, Bruker AXS Inc., Madison, WI, 2000 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- J. L. Zhu, Z. Xu, Y. Yang and L. Xu, Chem. Commun., 2019, 55, 6629–6671 RSC.
- C. Fennelly and R. K. Amaravadi, Methods Mol. Biol., 2017, 1594, 293–308 CrossRef CAS.
- K. Öllinger and H. Appelqvist, Methods Mol. Biol., 2017, 1594, 179–189 CrossRef.
- T. Miyayama and M. Matsuoka, J. Occup. Med. Toxicol., 2016, 11, 1–6 CrossRef.
- W. A. Wani, S. Prashar, S. Shreaz and S. Gómez-Ruiz, Coord. Chem. Rev., 2016, 312, 67–98 CrossRef CAS.
- K. Bruun and C. Hille, Sci. Rep., 2019, 9, 10504 CrossRef.
- A. Moreno, J. SantoDomingo, R. I. Fonteriz, C. D. Lobatón, M. Montero and J. Alvarez, J. Struct. Biol., 2016, 172, 261–269 CrossRef.
- X. Lou, M. Zhang, Z. Zhao, X. Min, A. Hakeem, F. Huang, P. Gao, F. Xia and B. Z. Tang, J. Mater. Chem. B, 2016, 4, 5412–5417 RSC.
- X. Lou, M. Zhang, Z. Zhao, X. Min, A. Hakeem, F. Huang, P. Gao, F. Xia and B. Z. Tang, J. Mater. Chem. B, 2016, 4, 5412–5417 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: UV-VIS spectroscopy, FTIR spectroscopy, ESI-MS spectroscopy, ORTEP diagram of complex, powder XRD, dynamic light scattering, SEM, TEM, confocal images. CCDC 2006521-2006523. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0bm01328e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |

 a,
Somali
Mukherjee‡
a,
Somali
Mukherjee‡