
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heparin-based, injectable microcarriers for controlled delivery of interleukin-13 to the brain†
Lucas
Schirmer
a,
Chloé
Hoornaert
b,
Debbie
Le Blon
bc,
Dimitri
Eigel
a,
Catia
Neto
d,
Mark
Gumbleton
d,
Petra B.
Welzel
a,
Anne E.
Rosser
ef,
Carsten
Werner
 ag,
Peter
Ponsaerts
bc and
Ben
Newland
*ad
ag,
Peter
Ponsaerts
bc and
Ben
Newland
*ad
aLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Str. 6, 01069 Dresden, Germany. E-mail: newlandb@cardiff.ac.uk
bLaboratory of Experimental Hematology, University of Antwerp, Antwerp, Belgium
cVaccine and Infectious Disease Institute, University of Antwerp, Antwerp, Belgium
dSchool of Pharmacy and Pharmaceutical Sciences, Cardiff University, CF10 3NB, Cardiff, UK
eBrain Repair Group, School of Biosciences, Cardiff University, Cardiff, CF10 3AX Wales, UK
fNeuroscience and Mental Health Institute and B.R.A.I.N unit. Cardiff University School of Medicine, Hadyn Ellis Building, Maindy Road, CF24 4HQ Cardiff, UK
gTechnische Universität Dresden, Center for Regenerative Therapies Dresden, Fetscherstr. 105, 01307 Dresden, Germany
First published on 1st September 2020
Abstract
Interleukin-13 (IL-13) drives cells of myeloid origin towards a more anti-inflammatory phenotype, but delivery to the brain remains problematic. Herein, we show that heparin-based cryogel microcarriers load high amounts of IL-13, releasing it slowly. Intra-striatal injection of loaded microcarriers caused local up-regulation of ARG1 in myeloid cells for pro-regenerative immunomodulation in the brain.
In the event of damage to the central nervous system (CNS), pro-inflammatory immune cells infiltrate the injured tissue to fend off infections and clear cellular debris. The resulting inflammation can add to the detrimental effects of a variety of different neuroinflammatory conditions, including ischemic stroke,1 multiple sclerosis,2,3 spinal cord injury,4 and epilepsy.5
Microglia, the resident immune cells of the CNS, adapt to these pathological changes in their environment by altering their phenotype, morphology, and their functions; towards the inflammatory M1 polarization state. Similar to macrophages, which can also be found in the CNS parenchyma, they can exist in a range of phenotypes that orchestrate the immune response in the region.6 However, alternative activation of microglia and macrophages can change their phenotype towards a regenerative polarization state (M2) and thus promote tissue regeneration. As major players in the regulation of inflammation/tissue repair, these regenerative microglia and macrophages, offer a promising target for therapeutic intervention in neuroinflammatory diseases.
One strategy to shift the polarization balance, is the administration of the known M2-inducing cytokine, interleukin-13 (IL-13).2,3 This anti-inflammatory cytokine, produced predominantly by Th2 lymphocytes, can inhibit the secretion of pro-inflammatory signaling mediators such as IL-1β, IL-6, IL-12, and TNF-α, while enhancing the expression of the mannose receptor and MHC II molecules.7 In addition, IL-13 has been shown to suppress the infiltration of inflammatory cells and to decrease axonal loss.8 Previous studies have demonstrated the potential of IL-13-based immunomodulation of microglia/macrophages towards an anti-inflammatory/pro-regenerative phenotype in the rodent CNS, which might be utilized as a promising therapeutic strategy for a range of inflammatory diseases such as multiple sclerosis, spinal cord injury, and stroke.1,2,4,5,9 Despite this cytokine's pronounced therapeutic potential, delivery to the CNS so far remained challenging.
The major limitation lies in the low long-term stability of most cytokines due to degradation and proteolysis under physiological conditions,10 which requires some form of continuous delivery for effective therapeutic application. Implantable infusion catheters have been developed for direct, sustained delivery of neurotrophic factors to the brain of Parkinson's patients.11 However, their application is limited by the increased risk of infections and other possible side effects such as hemorrhages or neurological complications.12
Besides the direct delivery of protein therapeutics, increased levels of the desired protein in the brain can also be achieved through viral vector-mediated overexpression or transplantation of cells engineered to secrete the therapeutic of interest.1,2,4,5,9 While gene-based delivery has been shown to be effective in supplying IL-13 to the target region, these methods often suffer from low transfection efficiency, variable durability of gene expression, possible mutations due to gene integration, and, in some cases, adverse immune reactions.13–15 As a viable alternative, an injectable protein delivery system that allows local sustained release of IL-13 could provide a less invasive and safer approach to modulate the immune response within a specific region of the brain. To the best of our knowledge, no delivery device for sustained IL-13 release has yet been developed.
Biomaterial-based approaches, such as encapsulation of proteins within polymer microparticles, represent a promising strategy for protein delivery to the brain.16 Polymers such as poly (lactic-co-glycolic) acid (PLGA) have been previously employed to release glial cell line-derived neurotrophic factor (GDNF) to the rodent and primate brain for therapeutic applications in Parkinson's disease.17,18 A drawback of most microencapsulation approaches is the limited control over payload release, which is often characterized by an initial diffusion-controlled burst release phase within the first day.18 Another problem associated with microencapsulation arises due to low protein stability within the microparticle, resulting in a loss in protein bioactivity.19 Thus, many microparticles are less than ideal vehicles for the therapeutic delivery of signaling mediators such as IL-13.19,20
As a viable alternative, soft biohybrid hydrogels containing glycosaminoglycans (GAGs) have been extensively used in order to biomimetically emulate functions of the extracellular matrix.21 Through their negative charge, sulfated GAGs such as heparin can interact with a range of growth factors, chemokines, and cytokines. Positively charged binding sites found on the protein's surface allow for electrostatic complexation and stabilization against proteolytic or thermal degradation. Incorporation of heparin into hydrogel networks, therefore, becomes a highly appealing strategy for the loading and sustained delivery of therapeutic proteins.22–28
Here, as an alternative to conventional hydrogels, we explore heparin-based macroporous cryogel microparticles (microcarriers) for sustained delivery of IL-13 into the brain. Macroporous cryogels have a unique sponge-like structure, which gives them several advantages over other biomaterials. Their compressibility, and their ability to reshape to their original size afterward, make them ideal for micro-invasive injection where they conform to the void space.30,31 Furthermore, their very high surface area to volume ratio facilitates electrostatic loading with proteins or other drugs.29
The presented study set out to prove whether injectable microscale hydrogel scaffolds, containing heparin as a building block and affinity center, could act as a sustained delivery device to polarize macrophages and microglia towards the pro-regenerative M2 phenotype in the brain. Cryogel microcarriers were thus prepared, characterized, and optimized for their interaction and release of IL-13. The immunomodulatory effects of IL-13 functionalized microcarriers were then assessed: first on bone marrow-derived macrophages in vitro and then in the murine brain in vivo (Fig. 1).
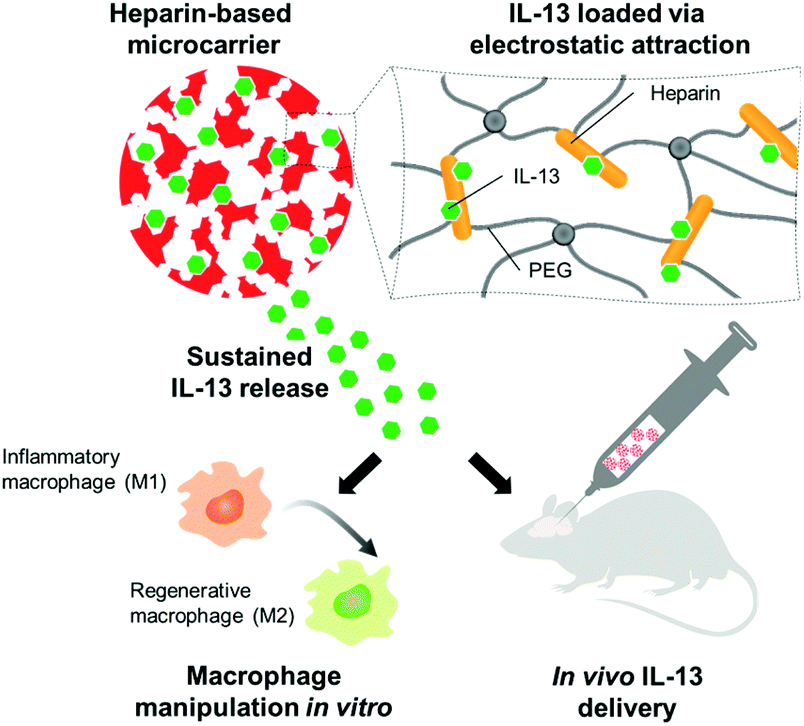 | ||
| Fig. 1 Schematic overview of the experimental approach. Heparin-based microcarriers were used to load and release IL-13 in order to change the phenotype of macrophages in vitro and induce a pro-regenerative phenotype in macrophages/microglia in the mouse brain. | ||
Design and characteristics of cryogels
Macroporous cryogels were formed using water as a porogen, by combining an emulsion technique with cryogelation of the aqueous heparin and poly(ethylene glycol) precursor solution at subzero temperatures.32 Under these conditions, the polymerization occurs in the non-frozen microphase between the ice crystals, creating robust struts and a porous structure.33Fig. 2 shows the macroporous structure of the microcarriers in the dry state (scanning electron microscopy image, Fig. 2A) and in the PBS swollen state (confocal laser scanning fluorescence microscopy image, Fig. 2B). The cross-section obtained via a single z-plane image (ESI Fig. S1†) shows that the microcarrier consists of thin struts and macropores and that the majority of the cross-sectional area is large open pores. Further analysis of the architecture through computational feature extraction and modeling demonstrated the high interconnectivity of the pores throughout the hydrogel matrix (Fig. 2C). Characterization of the microcarriers using confocal microscopy showed an average diameter of 321 μm (±131 μm) (Fig. 2D) and pore size of 23.4 μm (±11.3 μm) (Fig. 2E).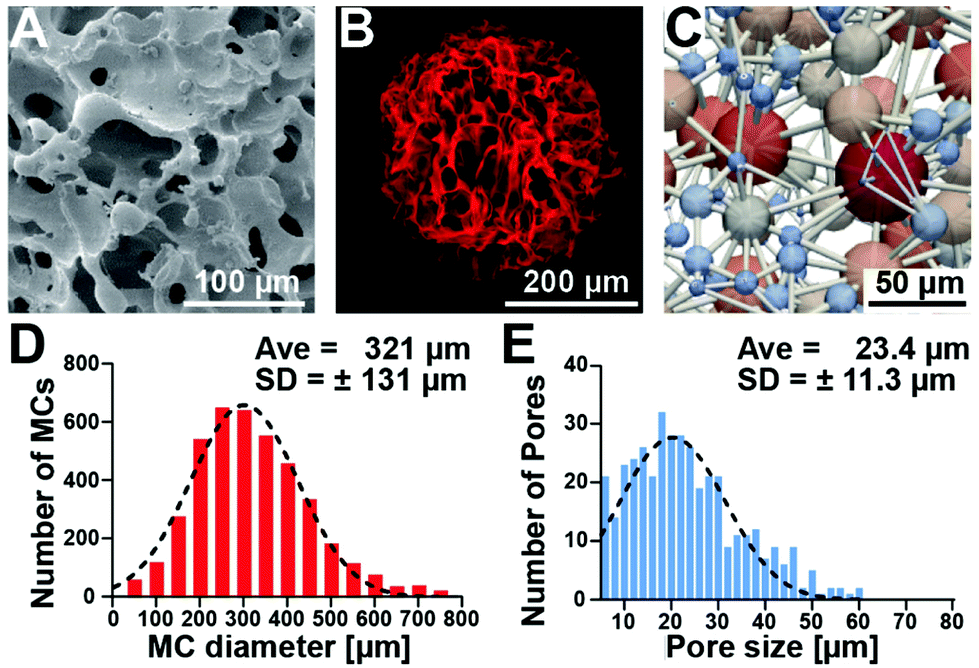 | ||
| Fig. 2 Microcarriers have a macroporous structure. (A) Representative scanning electron microscope image of a dry microcarrier. (B) Representative 40 μm z-projection (z-stack) of an ATTO 647 labeled microcarrier in phosphate-buffered saline visualized by confocal microscopy. (C) Computationally generated model of a pore network within a microcarrier. (D) Size distribution of microcarriers. (E) Pore size distribution within the microcarriers. | ||
The macroporous structure of cryogels allows them to be compressed to a small volume fraction of their original size yet retain “shape memory” and re-form to their original size and shape when the deforming force is removed.29–31,34 Cryogel materials can adsorb large amounts of energy without experiencing a large increase in stress. Consequently, these rather soft materials are very tough: we previously showed that they do not even break at a compression strain of over 90%.29 This ability to compress and expand is useful for applications where biomaterial-assisted delivery of cells or therapeutics through a small cannula size is required (i.e., stereotactic injection into the central nervous system).35 We tested the ability of microcarriers to be injected through a 30 gauge needle or a glass capillary (internal diameters of 160 and 140 μm respectively). ESI Fig. S2† shows the compression of a single microcarrier (420 μm diameter) through the glass capillary (compression ratio of 66%) with no visible change in microcarrier structure after injection.
Biofunctionalization of cryogels with IL-13
While the heparin-binding affinity of other similar 4-helical interleukins such as IL-4 has been well reported,36–39 there is rather limited experimental data about the heparin-affinity of IL-13. In silico modelling of the interaction of heparin with IL-13, based on the work of Mottarella et al.40 and Kozakov et al.41 helped to identify multiple potentially heparin-binding domains as previously described by Mulloy et al.42 (Fig. 3A and ESI Video S1†).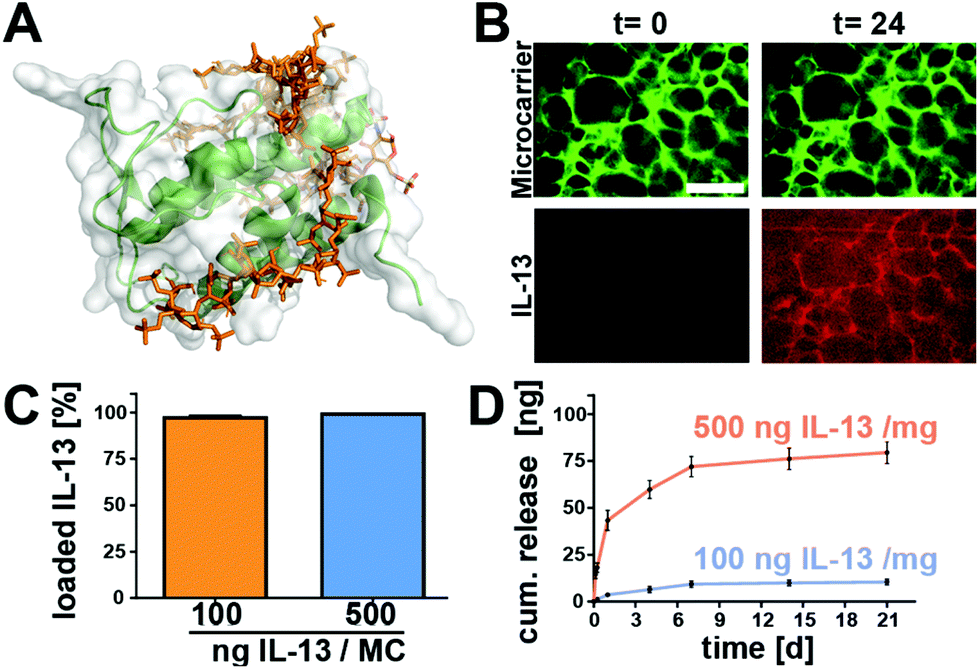 | ||
| Fig. 3 Heparin-based microcarriers bind and release IL-13. (A) Representative image of the in silico modeling of the electrostatic interaction of IL-13 (green) with the heparin biopolymer (orange). (B) Confocal laser microscope images of microcarriers (green) which take up IL-13 labeled with Atto-647 (red) within 24 hours. Scale bars = 100 μm. (C) For 100 or 500 ng of IL-13 per mg of microcarrier in the loading solution, 97.9% and 99.94% of the IL-13 were bound to the microcarriers, respectively. (D) IL-13 was released for up to three weeks (longest time tested) (n = 3 for all experiments and error bars represent ± standard deviation). | ||
The computational results were confirmed by the results of the IL-13 loading experiments, wherein microcarriers where incubated with either 100 ng or 500 ng IL-13. Analysis of the supernatants after 24 h revealed that at both loading concentrations, the microcarriers were able to take up nearly all of the IL-13 out of the solution, i.e., >95% of the IL-13 was bound to the microcarriers (Fig. 3C). Further analysis of the IL-13 loading, utilizing Atto-647 labeled protein, showed an accumulation of the IL-13 within the hydrogel matrix of the microcarriers (Fig. 3B). IL-13 fluorescence accumulated within the cryogel struts until reaching a maximum after 13 h (ESI Fig. S3†). Due to their strong affinity for IL-13, the microcarriers could be loaded with high amounts of IL-13 (a maximum of 500 ng mg−1 of microcarrier was tested) and the release of it into a buffer solution containing 1% bovine serum albumin was therefore expected to be slow and sustained.
Fig. 3D shows that IL-13 was indeed released without an initial burst for at least 21 days (longest time tested). For microcarriers loaded with 100 ng of IL-13 per mg of microcarrier, 9.3% of the IL-13 was released by day 7, continuing to 10.4% by day 21 (9.3 ng and 10.4 ng released respectively). The 500 ng mg−1 of microcarrier group released 12% of the IL-13 by day 7 and 15.9% by day 21 (71.9 ng and 79.5 ng released respectively). Although the chosen set-up does not fully recapitulate the complex in vivo environment (where multiple blood components, extracellular fluid, and other ECM components may competitively bind to the heparin to displace the IL-13), it gives us an indication that heparin-based cryogel microcarriers have the potential for therapeutic sustained IL-13 delivery. With up to 85% of the IL-13 remaining on the microcarriers, further release can be envisaged through displacement by extracellular molecules in vivo. By utilizing the strong affinity of IL-13 via binding sites on the AB loop and helix D42 to extracellular matrix glycosaminoglycans, such as heparin, a controlled, sustained release of IL-13 from the scaffold over several weeks could be achieved. Furthermore, protection of the protein load by the heparin-based hydrogel against degradation has been shown previously for the structurally similar 4-helical protein IL-4.43
Immunomodulatory activity of IL-13 functionalized cryogels on bone-marrow derived macrophages
In vitro tests with mouse bone marrow-derived macrophages were performed to assess if IL-13 released from the microcarriers could induce polarization of macrophages towards the alternatively activated (M2) state. It has been well established that they respond to IL-13 in vitro.44 Furthermore, it has been well-characterized that alternative M2 activation of murine macrophages induces the upregulation of Arg1 and Ym1 signature genes.45Fig. 4A shows a schematic depiction of the process of loading 1 mg microcarriers in a solution (200 μL) containing either 100 ng or 500 ng of IL-13, followed by the washing step and placement of the microcarriers in culture with the macrophages. Subsequent gene expression analysis showed that IL-13 loaded microcarriers (at both loading concentrations) caused a major upregulation of the M2 marker genes Arg1 (Fig. 4B) and Ym1 (Fig. 4C). The expression levels were similar to that obtained when IL-13 was directly added into the macrophage medium as a positive control (IL-13 in solution group). Control microcarriers that were not loaded with IL-13 (empty microcarrier group) caused no increase in the expression of either Arg1 or Ym1, indicating that the microcarriers themselves do not affect the expression of these genes. As this cell culture experiment was carried out for three days, approximately 5–10 ng of IL-13 should have been released into the macrophage medium from the microcarriers loaded with 100 ng mg−1 (deduced from the release experiment data, Fig. 3D). This was below the concentration of IL-13 in the soluble IL-13 positive control group (10 ng mL−1) but was still enough to induce strong upregulation of both genes.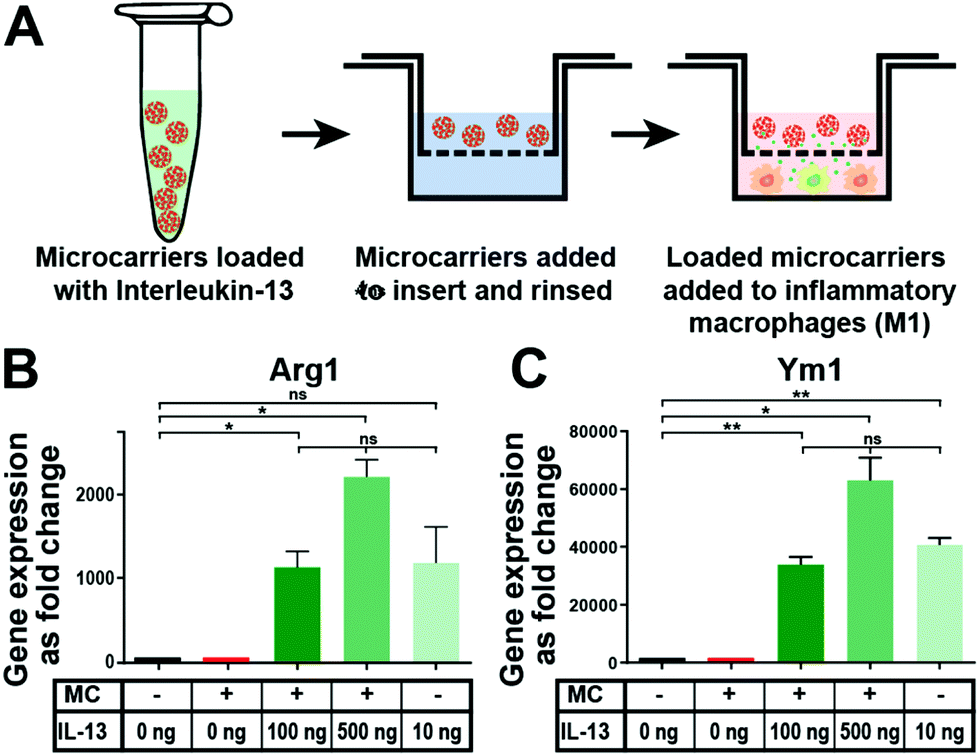 | ||
| Fig. 4 IL-13 released from microcarriers induces alternative activation of macrophages in vitro. (A) Schematic depiction of the experimental process of microcarrier loading with IL-13 and testing their effect on inflammatory macrophages. Gene expression analysis showed that IL-13 loaded microcarriers caused an upregulation of alternative activation polarization markers Arg1 (B) and Ym1 (C) (n = 3, one-way ANOVA with Tukey's post-hoc analysis ns = no significant difference, * represents a P-value ≤ 0.05, ** represents a P-value ≤ 0.01). | ||
Immunomodulatory activity of IL-13 functionalized cryogels in the brain
A delivery system that slowly releases IL-13 in the CNS could be utilized for a variety of applications. Since any injection into the brain results in a needle stick injury, whereby macrophages and microglia invade the injection tract (an event which is seen even when injecting only sterile saline46), such a sustained-release system could be used as an adjuvant for regenerative medicine and cell transplantation strategies. The microglial/macrophage response associated with the stereotactic injection is typically pro-inflammatory in nature,9,47 adding a greater burden to the brain region undergoing treatment. In the example of cell replacement therapies, the host brain can become a hostile environment for transplanted cells,48,49 and the rapid death of grafted cells post-transplantation may, in part, be due to this adverse response.50–52 The microcarrier-based IL-13 delivery system may therefore provide a suitable platform technology for modulating the host response to implanted cells, viral vectors, or other therapeutics. To this end, an in vivo study was performed whereby either empty microcarriers or IL-13 loaded microcarriers were stereotactically administered to the adult mouse striatum.The host response to injection of empty versus IL-13 loaded microcarriers was analyzed and compared with the contralateral side of the brain that received no injection (Fig. 5A, microcarrier regions outlined with a dashed line). GFAP+ astrocytes (astrocytic scar) migrated to the border but did not really infiltrate either empty or IL-13 loaded microcarriers (Fig. 5B). In contrast, IBA1+ reactive microglia could be observed surrounding and invading the microcarrier-injected area. This pattern of host response is very similar to that observed when grafting cells into the rodent brain, where the cell graft becomes encapsulated by an astrocytic scar and infiltrated and surrounded by cells of myeloid origin.51,53 Quantification of the GFAP+ and IBA1+ cells through immunohistochemistry is shown in Fig. 5C and D, respectively, where no significant difference in the area of staining could be observed between empty and IL-13 loaded microcarriers. This illustrates that the IL-13 itself is not exacerbating the host glial scar or microglial response.
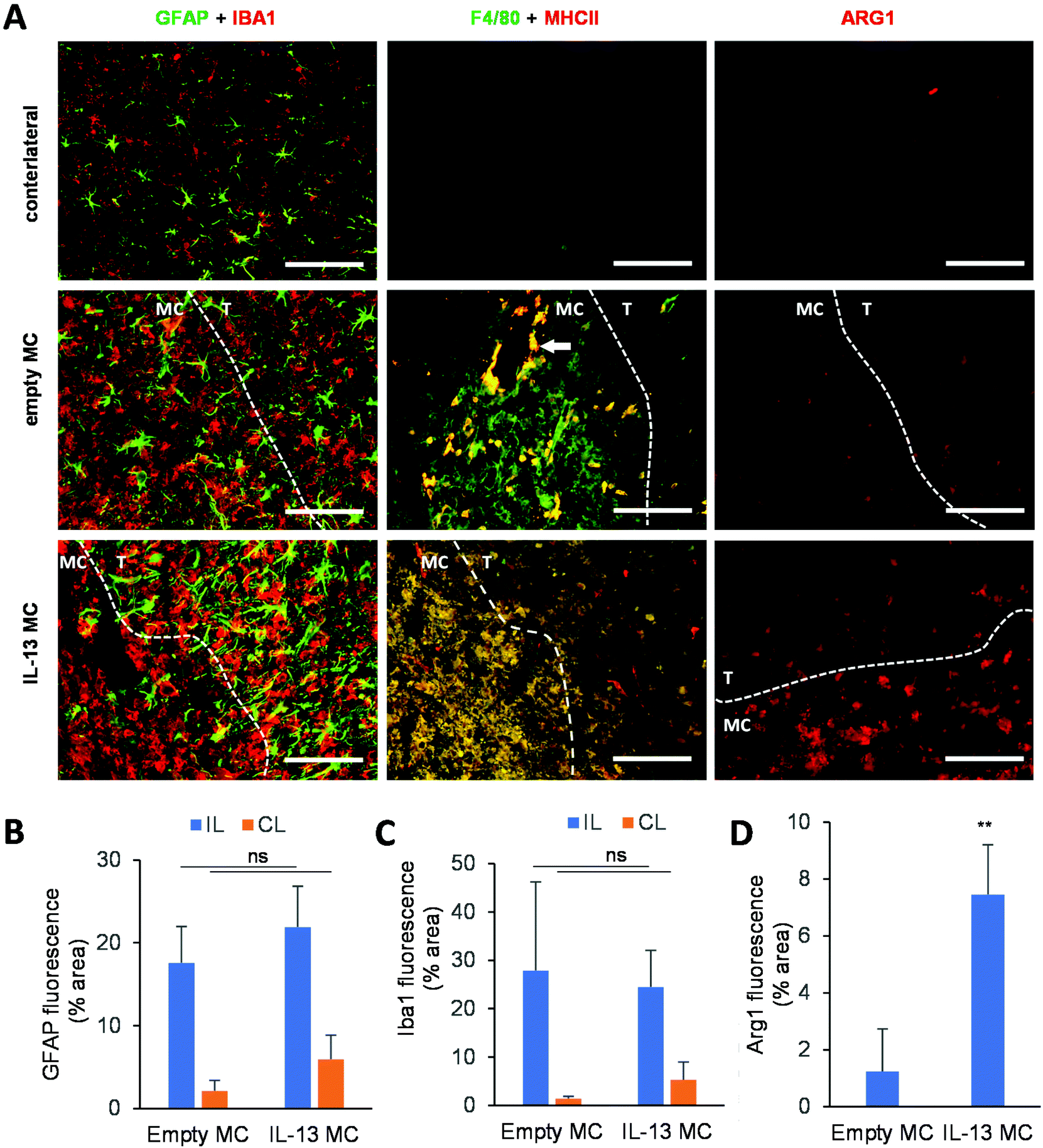 | ||
| Fig. 5 IL-13 released from microcarriers induces alternative activation of macrophages in vivo. Histological analyses were performed one week after in vivo delivery of empty microcarriers (empty MC) (n = 3) or IL-13-loaded microcarriers (IL-13 MC) (n = 4). (A) Immunofluorescent staining for GFAP + astrocytes, IBA1 + myeloid cells, F4/80 + MHCII− activated microglia, F4/80 + MHCII+ activated macrophages, and ARG1 + M2 polarized myeloid cells. MC injection area is outlined with a dashed line where MC = microcarrier, and T = brain tissue. The white arrow indicates the location of a blood vessel, scale bars = 100 μm. (B–D) Quantification of the relative surface area that stained positive for the markers GFAP (B), IBA1(C), or ARG1(D). Data points represent mean per condition (IL = ipsilateral, CL = contralateral), with error bars representing ± standard deviation. A Student's t-test was performed to analyze statistically significant differences between empty and IL-13 loaded microcarriers (ns = no significant difference, ** represents a P-value ≤ 0.01). | ||
To analyze the microglia/macrophage infiltration of the microcarrier injection site, F4/80 immunohistochemical analysis (general myeloid activation marker) was performed in conjunction with major histocompatibility class II (MHC II). The injection sites of empty microcarrier were predominantly infiltrated by F4/80+ cells, with just a small fraction of the cells showing dual F4/80 and MHC II staining. Interestingly, F4/80+ MHC II+ double-positive cells were restricted to regions surrounding blood vessels, indicating their monocytic origin. IL-13 loaded microcarriers, on the other hand, displayed an infiltration by F4/80+ MHC II+ double-positive macrophages. This latter infiltration pattern is reminiscent of mesenchymal stem cell grafts, where IBA1+ MHCII+ macrophages predominantly infiltrate the cell graft while IBA1+ MHCII− resident microglia accumulate in the graft border.54 Furthermore, a significant fraction of immune cells that infiltrated the IL-13 loaded microcarrier injection sites, but not empty microcarrier sites, were positive for ARG1 expression. This suggests that the IL-13 released from the microcarriers is able to alternatively activate either the resident microglia or the peripheral macrophages. We have previously shown, via injection of cells engineered to overexpress IL-13, that the phenotypic shift towards the pro-regenerative ARG1+ M2a phenotype is predominantly observed in macrophages rather than microglia,4 hence confirming the monocytic origin of IBA1+ ARG1+ cells infiltrating the graft as well as the microglial origin of IBA1+ ARG1+ cells found in the injection site border.
With the emergence of high-throughput, high-precision techniques such as single-cell RNA sequencing, the high complexity and dynamism of microglial and macrophage phenotypes are becoming ever more apparent.55 Since these cells’ primary function consists of immune surveillance of their surroundings, they are well-adapted to respond to a plethora of local cues that convey information regarding potential infections and/or tissue damage. Among these, toll-like receptor ligands (e.g., LPS or dsRNA) and pro-inflammatory cytokines (e.g., IL1β and TNF) will cause a shift to a more inflammatory phenotype,56 whilst the Th2 cytokines IL-4, IL-10 and IL-13 have been shown to drive more anti-inflammatory phenotypes.57,58 It is therefore conceivable that these interleukins could be utilized to enhance the regenerative capacity of the central nervous system by inducing endogenous repair. Even so, the transient nature of the phenotypic shift would likely necessitate a constant supply of the cytokine of interest.
Poor protein stability in vivo due to proteolytic degradation has driven the development of infusion pumps, such as those used for continued growth factor delivery to the brain.11 Alternatively to direct protein infusion, both gene and cell therapeutic approaches could be considered. We previously showed that injection of IL-13 encoding lentiviral vectors, as well as transplantation of IL-13 expressing carrier cells in the CNS, resulted in a local and sustained production of IL-13.1,2,4,9,59 Despite the advantages of both approaches, viral strategies still pose a safety concern (resulting from excessive TLR activation of CNS microglia/macrophages in response to the viral particles) whereas cell-mediated approaches are not without drawbacks, in particular graft rejection or uncontrolled cell growth/death.1
We herein proposed an alternative approach, showing that microscale biomaterial constructs give a slow and sustained release of IL-13 and cause ARG1 over-expression for at least seven days following intra-striatal injection. Thus, we could demonstrate that biomaterial-based delivery systems can be used to modulate the host inflammatory response, which could impact future surgical interventions in the brain without the need for cell (and subsequent immunosuppression) or viral gene delivery through a single infusion for relatively acute mediation.
Further work is warranted to determine the loaded cryogel storage capabilities, the full duration of IL-13 release, long-term host responses to the microcarriers (with and without the payload), and whether similar results can be achieved with other immunomodulatory factors such as IL-4.
Conclusions
Herein, we report on the synthesis of microscale cryogels (microcarriers) comprised of heparin and four-arm PEG, and on the analysis of their suitability as a sustained delivery system for the immunomodulatory cytokine IL-13. 1 mg of these microcarriers could be loaded with at least 500 ng of IL-13 and released 13% of the cytokine over a period of three weeks without a significant burst release in the first few hours. IL-13 functionalized microcarriers caused overexpression of ARG1 (anti-inflammatory phenotype marker) both in macrophages cultured in vitro, and in microglia/macrophages of the brain in vivo. This study proves the concept that injectable cryogel microcarriers, utilizing heparin to non-covalently complex IL-13, can deliver the cytokine to the CNS and thus modulate the local immune response towards a pro-regenerative M2 polarization.Author contributions
B.N. conceived the project. B.N. and L.S. designed the material characterization and in vitro studies. L.S. performed all the in vitro experiments; B.N. and D.E. performed the materials characterization. C.H. and D.L.B. planned, performed, and analyzed the in vivo study. B.N., L.S., C.H., and P.B.W. wrote the initial manuscript draft, and all authors discussed the results and contributed to the manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dagmar Pette for her support and for preparing the materials used in these studies. Funding: This work was funded by the Wellcome Trust (Sir Henry Wellcome Postdoctoral Fellowship (BN) and the Deutsche Forschungsgemeinschaft – Project number 320041273.Notes and references
- S. Hamzei Taj, D. Le Blon, C. Hoornaert, J. Daans, A. Quarta, J. Praet, A. Van der Linden, P. Ponsaerts and M. Hoehn, J. Neuroinflammation, 2018, 15, 174 CrossRef.
- C. Guglielmetti, D. Le Blon, E. Santermans, A. Salas-Perdomo, J. Daans, N. De Vocht, D. Shah, C. Hoornaert, J. Praet, J. Peerlings, F. Kara, C. Bigot, Z. Mai, H. Goossens, N. Hens, S. Hendrix, M. Verhoye, A. M. Planas, Z. Berneman, A. van der Linden and P. Ponsaerts, Glia, 2016, 64, 2181–2200 CrossRef.
- V. E. Miron, A. Boyd, J.-W. Zhao, T. J. Yuen, J. M. Ruckh, J. L. Shadrach, P. van Wijngaarden, A. J. Wagers, A. Williams, R. J. M. Franklin and C. Ffrench-Constant, Nat. Neurosci., 2013, 16, 1211–1218 CrossRef.
- D. Dooley, E. Lemmens, T. Vangansewinkel, D. Le Blon, C. Hoornaert, P. Ponsaerts and S. Hendrix, Stem Cell Rep., 2016, 7, 1099–1115 CrossRef.
- I. Ali, S. Aertgeerts, D. Le Blon, D. Bertoglio, C. Hoornaert, P. Ponsaerts and S. Dedeurwaerdere, Epilepsia, 2017, 58, 1063–1072 CrossRef.
- Q. Li and B. A. Barres, Nat. Rev. Immunol., 2018, 18, 225–242 CrossRef CAS.
- K. M. DeFife, C. R. Jenney, A. K. McNally, E. Colton and J. M. Anderson, J. Immunol., 1997, 158, 3385–3390 CAS.
- J. Ochoa-Repáraz, A. Rynda, M. A. Ascón, X. Yang, I. Kochetkova, C. Riccardi, G. Callis, T. Trunkle and D. W. Pascual, J. Immunol., 2008, 181, 954–968 CrossRef.
- D. Le Blon, C. Guglielmetti, C. Hoornaert, A. Quarta, J. Daans, D. Dooley, E. Lemmens, J. Praet, N. De Vocht, K. Reekmans, E. Santermans, N. Hens, H. Goossens, M. Verhoye, A. Van der Linden, Z. Berneman, S. Hendrix and P. Ponsaerts, J. Neuroinflammation, 2016, 13, 288 CrossRef.
- R. Pearlman and Y. J. Wang, Formulation, Characterization, and Stability of Protein Drugs: Case Histories, Springer US, Boston, MA, 1st edn, 2002, vol. 9 Search PubMed.
- A. Whone, M. Luz, M. Boca, M. Woolley, L. Mooney, S. Dharia, J. Broadfoot, D. Cronin, C. Schroers, N. U. Barua, L. Longpre, C. L. Barclay, C. Boiko, G. A. Johnson, H. C. Fibiger, R. Harrison, O. Lewis, G. Pritchard, M. Howell, C. Irving, D. Johnson, S. Kinch, C. Marshall, A. D. Lawrence, S. Blinder, V. Sossi, A. J. Stoessl, P. Skinner, E. Mohr and S. S. Gill, Brain, 2019, 142, 512–525 CrossRef.
- T. J. Kaufmann, J. Huston, J. N. Mandrekar, C. D. Schleck, K. R. Thielen and D. F. Kallmes, Radiology, 2007, 243, 812–819 CrossRef.
- T. N. Demidova-Rice, M. R. Hamblin and I. M. Herman, Adv. Skin Wound Care, 2012, 25, 349–370 CrossRef.
- J. Y. Sun, V. Anand-Jawa, S. Chatterjee and K. K. Wong, Gene Ther., 2003, 10, 964–976 CrossRef CAS.
- M. E. Gore, Gene Ther., 2003, 10, 4–4 CrossRef CAS.
- B. Newland, H. Newland, C. Werner, A. Rosser and W. Wang, Prog. Polym. Sci., 2015, 44, 79–112 CrossRef CAS.
- E. Garbayo, C. N. Montero-Menei, E. Ansorena, J. L. Lanciego, M. S. Aymerich and M. J. Blanco-Prieto, J. Controlled Release, 2009, 135, 119–126 CrossRef CAS.
- E. Garbayo, E. Ansorena, H. Lana, M. del M. Carmona-Abellan, I. Marcilla, J. L. Lanciego, M. R. Luquin and M. J. Blanco-Prieto, Biomaterials, 2016, 110, 11–23 CrossRef CAS.
- M. van de Weert, W. E. Hennink and W. Jiskoot, Pharm. Res., 2000, 17, 1159–1167 CrossRef CAS.
- S. C. Yadav, A. Kumari and R. Yadav, Peptides, 2011, 32, 173–187 CrossRef CAS.
- U. Freudenberg, Y. Liang, K. L. Kiick and C. Werner, Adv. Mater., 2016, 28, 8861–8891 CrossRef CAS.
- O. Jeon, C. Powell, L. D. Solorio, M. D. Krebs and E. Alsberg, J. Controlled Release, 2011, 154, 258–266 CrossRef CAS.
- T. Nie, A. Baldwin, N. Yamaguchi and K. L. Kiick, J. Controlled Release, 2007, 122, 287–296 CrossRef CAS.
- S. E. Sakiyama-Elbert and J. A. Hubbell, J. Controlled Release, 2000, 69, 149–158 CrossRef CAS.
- J. Schroeder-Tefft, H. Bentz and T. Estridge, J. Controlled Release, 1997, 49, 291–298 CrossRef CAS.
- A. Zieris, K. Chwalek, S. Prokoph, K. R. Levental, P. B. Welzel, U. Freudenberg and C. Werner, J. Controlled Release, 2011, 156, 28–36 CrossRef CAS.
- U. Freudenberg, A. Zieris, K. Chwalek, M. V. Tsurkan, M. F. Maitz, P. Atallah, K. R. Levental, S. A. Eming and C. Werner, J. Controlled Release, 2015, 220, 79–88 CrossRef CAS.
- S. T. Koshy, D. K. Y. Zhang, J. M. Grolman, A. G. Stafford and D. J. Mooney, Acta Biomater., 2018, 65, 36–43 CrossRef CAS.
- B. Newland, P. B. Welzel, H. Newland, C. Renneberg, P. Kolar, M. Tsurkan, A. Rosser, U. Freudenberg and C. Werner, Small, 2015, 11, 5047–5053 CrossRef CAS.
- S. A. Bencherif, R. W. Sands, D. Bhatta, P. Arany, C. S. Verbeke, D. A. Edwards and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19590–19595 CrossRef CAS.
- A. Béduer, T. Braschler, O. Peric, G. E. Fantner, S. Mosser, P. C. Fraering, S. Benchérif, D. J. Mooney and P. Renaud, Adv. Healthcare Mater., 2015, 4, 301–312 CrossRef.
- K. R. Hixon, T. Lu and S. A. Sell, Acta Biomater., 2017, 62, 29–41 CrossRef CAS.
- P. B. Welzel, J. Friedrichs, M. Grimmer, S. Vogler, U. Freudenberg and C. Werner, Adv. Healthcare Mater., 2014, 3, 1849–1853 CrossRef CAS.
- S. A. Bencherif, R. Warren Sands, O. A. Ali, W. A. Li, S. A. Lewin, T. M. Braschler, T.-Y. Shih, C. S. Verbeke, D. Bhatta, G. Dranoff and D. J. Mooney, Nat. Commun., 2015, 6, 7556 CrossRef CAS.
- B. Newland, S. B. Dunnett and E. Dowd, Drug Discovery Today, 2016, 21, 1313–1320 CrossRef CAS.
- C. A. Jones, K. A. Williams, J. J. Finlay-Jones and P. H. Hart, Biol. Reprod., 1995, 52, 839–847 CrossRef CAS.
- H. Lortat-Jacob, P. Garrone, J. Banchereau and J.-A. Grimaud, Cytokine, 1997, 9, 101–105 CrossRef CAS.
- E. den Dekker, S. Grefte, T. Huijs, G. B. ten Dam, E. M. M. Versteeg, L. C. J. van den Berk, B. A. Bladergroen, T. H. van Kuppevelt, C. G. Figdor and R. Torensma, J. Immunol., 2008, 180, 3680–3688 CrossRef CAS.
- L. Schirmer, P. Atallah, C. Werner and U. Freudenberg, Adv. Healthcare Mater., 2016, 5, 3157–3164 CrossRef CAS.
- S. E. Mottarella, D. Beglov, N. Beglova, M. A. Nugent, D. Kozakov and S. Vajda, J. Chem. Inf. Model., 2014, 54, 2068–2078 CrossRef CAS.
- D. Kozakov, R. Brenke, S. R. Comeau and S. Vajda, Proteins, 2006, 65, 392–406 CrossRef CAS.
- B. Mulloy and M. J. Forster, Mol. Simul., 2008, 34, 481–489 CrossRef CAS.
- L. Schirmer, P. Atallah, C. Werner and U. Freudenberg, Adv. Healthcare Mater., 2016, 5, 3157–3164 CrossRef CAS.
- T. M. Doherty, R. Kastelein, S. Menon, S. Andrade and R. L. Coffman, J. Immunol., 1993, 151, 7151–7160 CAS.
- S. Gordon and F. O. Martinez, Immunity, 2010, 32, 593–604 CrossRef CAS.
- J. R. Detrez, H. Maurin, K. Van Kolen, R. Willems, J. Colombelli, B. Lechat, B. Roucourt, F. Van Leuven, S. Baatout, P. Larsen, R. Nuydens, J.-P. Timmermans and W. H. De Vos, Neurobiol. Dis., 2019, 127, 398–409 CrossRef CAS.
- N. De Vocht, D. Lin, J. Praet, C. Hoornaert, K. Reekmans, D. Le Blon, J. Daans, P. Pauwels, H. Goossens, N. Hens, Z. Berneman, A. Van der Linden and P. Ponsaerts, Immunobiology, 2013, 218, 696–705 CrossRef CAS.
- V. H. Roberton, A. E. Evans, D. J. Harrison, S. V. Precious, S. B. Dunnett, C. M. Kelly and A. E. Rosser, NeuroReport, 2013, 24, 1010–1015 CrossRef.
- C. J. Hoornaert, D. Le Blon, A. Quarta, J. Daans, H. Goossens, Z. Berneman and P. Ponsaerts, Stem Cells Transl. Med., 2017, 6, 1434–1441 CrossRef.
- D. B. Hoban, B. Newland, T. C. Moloney, L. Howard, A. Pandit and E. Dowd, Biomaterials, 2013, 34, 9420–9429 CrossRef CAS.
- T. C. Moloney, P. Dockery, A. J. Windebank, F. P. Barry, L. Howard and E. Dowd, Neurorehabil. Neural Repair, 2010, 24, 645–656 CrossRef.
- K. Reekmans, N. De Vocht, J. Praet, E. Fransen, D. Le Blon, C. Hoornaert, J. Daans, H. Goossens, A. Van der Linden, Z. Berneman and P. Ponsaerts, Stem Cell Res. Ther., 2012, 3, 56 CrossRef.
- C. W. Olanow, M. Savolainen, Y. Chu, G. M. Halliday and J. H. Kordower, Brain, 2019, 142, 1690–1700 CrossRef.
- D. Le Blon, C. Hoornaert, J. Daans, E. Santermans, N. Hens, H. Goossens, Z. Berneman and P. Ponsaerts, Immunol. Cell Biol., 2014, 92, 650–658 CrossRef CAS.
- T. R. Hammond, C. Dufort, L. Dissing-Olesen, S. Giera, A. Young, A. Wysoker, A. J. Walker, F. Gergits, M. Segel, J. Nemesh, S. E. Marsh, A. Saunders, E. Macosko, F. Ginhoux, J. Chen, R. J. M. Franklin, X. Piao, S. A. McCarroll and B. Stevens, Immunity, 2019, 50, 253–271.e6 CrossRef CAS.
- N. Parameswaran and S. Patial, Crit. Rev. Eukaryotic Gene Expression, 2010, 20, 87–103 CrossRef CAS.
- I. Francos-Quijorna, J. Amo-Aparicio, A. Martinez-Muriana and R. López-Vales, Glia, 2016, 64, 2079–2092 CrossRef.
- X. Hu, R. K. Leak, Y. Shi, J. Suenaga, Y. Gao, P. Zheng and J. Chen, Nat. Rev. Neurol., 2015, 11, 56–64 CrossRef.
- C. J. Hoornaert, E. Luyckx, K. Reekmans, M. Dhainaut, C. Guglielmetti, D. Le Blon, D. Dooley, E. Fransen, J. Daans, L. Verbeeck, A. Quarta, N. De Vocht, E. Lemmens, H. Goossens, A. Van der Linden, V. D. Roobrouck, C. Verfaillie, S. Hendrix, M. Moser, Z. N. Berneman and P. Ponsaerts, Stem Cells, 2016, 34, 1971–1984 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01249a |
| This journal is © The Royal Society of Chemistry 2020 |