
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe biomaterial polyphosphate blocks stoichiometric binding of the SARS-CoV-2 S-protein to the cellular ACE2 receptor
Werner E. G.
Müller
 *a,
Meik
Neufurth
a,
Hadrian
Schepler
b,
Shunfeng
Wang
a,
Emad
Tolba
a,
Heinz C.
Schröder
a and
Xiaohong
Wang
*a
*a,
Meik
Neufurth
a,
Hadrian
Schepler
b,
Shunfeng
Wang
a,
Emad
Tolba
a,
Heinz C.
Schröder
a and
Xiaohong
Wang
*a
aERC Advanced Investigator Grant Research Group at the Institute for Physiological Chemistry, University Medical Center of the Johannes Gutenberg University, Duesbergweg 6, 55128 Mainz, Germany. E-mail: wmueller@uni-mainz.de; wang013@uni-mainz.de
bDepartment of Dermatology, University Clinic Mainz, Langenbeckstr. 1, 55131 Mainz, Germany
First published on 14th October 2020
Abstract
The effect of the polyanionic polymer of inorganic polyphosphate (polyP) involved in innate immunity on the binding of the receptor-binding domain (RBD) of the SARS-CoV-2 spike protein to the cellular ACE2 receptor was studied. The RBD surface comprises a basic amino acid stretch of four arginine residues which interact with the physiological polyP (polyP40) and polyP3. Subsequently, the interaction of RBD with ACE2 is sensitively inhibited. After the chemical modification of arginine, an increased inhibition by polyP, at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio (polyP:RBP), is measured already at 0.1 μg mL−1. Heparin was ineffective. The results suggest a potential therapeutic benefit of polyP against SARS-CoV-2 infection.
:1 molar ratio (polyP:RBP), is measured already at 0.1 μg mL−1. Heparin was ineffective. The results suggest a potential therapeutic benefit of polyP against SARS-CoV-2 infection.
Introduction
Innate immunity is an evolutionary old mechanism present in the whole animal kingdom and is the first line of defense of the host to prevent infection and attack of invading pathogens.1 In particular, during the initial fast phase of infection (about one week), the host depends on the innate immune system to control, e.g. viral replication without excessive and adverse damage of the infected tissue.2 In this phase, conserved viral/bacterial structures, known as pathogen associated molecular patterns, serve as ligands for the pattern recognition molecules of the host, such as the Toll-like receptors.3 After receptor sensing, downstream signaling pathways are activated, resulting in the elicitation of an effective antimicrobial response, including the formation of physical barriers, activation of epithelial and phagocytic cell enzymes, phagocytes, as well as inflammation-related serum proteins. In turn, surface and phagocyte granule antimicrobial peptides as well as cytokines and inflammatory mediators are distributed.4 Besides the innate immune cells, especially neutrophils, monocytes and macrophages/mast cells, the blood platelets guide the innate immune system.5 Platelets are the key cells involved in blood hemostasis and coagulation, but also function as active participants in innate immunity.6 Both the negatively charged polymers, inorganic polyphosphate (polyP) and heparin, which are synthesized in mast cells and/or blood platelets, are mediators or modulators as well as effector molecules in the innate immunity defense.4,7 Heparin amplifies the network elicited by humoral-based proteins and/or cell-surface receptors and provides co-stimulatory signals for T-cells.8 This polysaccharide is released into the vascular system at the sites of tissue injury. PolyP as a member of the innate immune system is considered as an activator of coagulation and a suppressor of complement activation. The polymer is exported from platelets in response to a series of stimuli, such as prothrombotic factors or also after an interaction with collagen or after sensitization of receptors with immune-receptor tyrosine based motifs.9In an earlier study, we described that polyP inhibits human immunodeficiency virus-1 (HIV-1) in vitro, most likely via binding to both the infected cells and HIV-1.10 Similarly, mannose-specific lectins were also found to be inhibitory against viral infection in vitro, both against HIV-111 and coronaviruses.12 Now during the search for compounds acting as potential inhibitors of SARS-CoV-2 infection that might gain therapeutic impact, we studied the effects of the natural polymers of polyP (reviewed in: ref. 13) and – in comparison – of heparin on the binding of the virus to the target cells. A model system was applied for these studies, a binding assay, in which the cellular receptor for SARS-CoV-2, the recombinant angiotensin-converting enzyme 2 (ACE2),14 was attached to the well plate and the labeled receptor-binding domain (RBD) of the SARS-CoV-2 spike (S)-protein was used as a detector molecule.15 The viral RBD16 is highly characteristic of SARS-CoV-2 (expected value of 2 e−1;66 PDB: 6 M0J_E). One feature of the RBD is prominent: the number and the arrangement of basic amino acids (aa) on the modeled surface of the RBD. The RBD is spanning the aa segment at the SARS-CoV-2 S-protein sequence14,16 and comprises a surplus of basic aa (with Arg [arginine] + Lys [lysine] + His [histidine]: 11 + 10 + 1) over acidic aa (Asp [aspartate] + Glu [glutamate]: 15). Therefore, heparins with their polyanionic surfaces have been proposed to interact with the S-protein of coronaviruses in general17 and with SARS-CoV-2 in particular.18 Moreover, it has been reported that heparin even if applied at a concentration of 100 μg mL−1, a concentration that is much higher than the normal physiological concentration of heparin in human plasma (1 to 5 μg mL−1),19 inhibits virus invasion.20,21 This conclusion is based upon surface plasmon resonance and circular dichroism spectroscopy. Recently it has been hypothesized, based on computational ligand docking experiments, that heparan sulfate interacts with the glycosaminoglycan-binding motif at the S1/S2 proteolytic cleavage site within the SARS-CoV-2 spike glycoprotein.22 In the present study, we could provide functional evidence that heparin has no effect on the interaction between the viral RBD and the ACE2 receptor.
Most of the basic aa of the SARS-CoV-2 RBD are arranged on the surface in a clustered pattern (Fig. 1), leaving a spacing between 3.8 and 5 Å. Arg-rich peptides are frequently interspersed in cell-penetrating peptides and also in intracellular binding proteins which are characterized by their strong binding affinity to their respective ligand. An example is the HIV-1 Tat protein that binds to the hair-looped structured viral TAR RNA and links this complex to the nuclear matrix.23 The nine aa long Tat peptide, consisting of the protein transduction domain, comprises six Arg, two Lys and only one polar/not charged aa (Gln), allowing penetration into cells.24,25
 | ||
| Fig. 1 Mapping of the peptide surfaces of RBD by a prediction of (A) amino acid clustering, (B) hydrophobicity and (C) electric charge distribution. The basic aa in (A) are highlighted in green (Arg [R] and Lys [K]) and the two acidic aa Asp [D] and Glu [E] in purple. The interphase between the RBD of the SARS-CoV-2 S-protein and the ACE2 is marked. In (B), the hydrophobicity distribution is highlighted from −4.5 (the index for Arg, the sign for not very hydrophobic) to +4.5 (hydrophobic). In (C), the electric charge distribution is shown, based on Poisson–Boltzmann calculations, and marked from −10 (more hydrophobic areas) to +10 (hydrophilic). The direction of the predicted polyP groove is indicated with a green bar and the aa pair Asp and Glu is highlighted with a purple bar. | ||
Interesting is the observation that pores formed from Lys-rich artificial β-barrels are anion selective, while Arg-rich pores are cation selective.26 This apparent contradiction has been explained by a binding of the Arg-segments to phosphate counterions that invert the internal charge from positive to negative, making the pores selective for cations.24 These electrostatic interactions between a basic epitope, comprising Arg residues, and an adjacent acidic epitope, containing a phosphorylated group, are strong.27 In turn, electrostatic interactions between Arg and phosphate are expected to combine from at least 4 Arg residues on the surface of the RBD (Fig. 1). The physiological polyP has been shown to be co-localized with basic aa in cellular vesicles, e.g. fungal vacuoles, in a 1:1 stoichiometric ratio.28 A strong binding to Arg in a peptide has also been reported for terminal sialic acid residues in glycoproteins like in the coronavirus attachment to the sialic acid receptors.29 A selective and strong binding of heparin to Arg- and Lys-rich domains has likewise been established.30
These results suggested that both the anionic polyP and the anionic heparin might interact with the Arg stretches on the surface of the RBD of the SARS-CoV-2 S-protein. An interaction of heparin with the RBD by a conformational change has been published.18 In the present study, the effect of both polyP and heparin on the interaction between SARS-CoV-2 and RBD has been investigated. The results show that only polyP but not heparin is capable of inhibiting the binding of RBD to the recombinant receptor. To pinpoint the effect of polyP on the RBD:ACE2 interaction to the aa level of RBD, Arg residues have been modified with 1,2-cyclohexanedione (CHD), which reacts specifically with the guanidino group of arginine at pH 8 to 9 in sodium borate buffer to derivatize arginine to N7,N8-(1,2-dihydroxycyclohex-1,2-ylene)-L-arginine [DHCH-Arg].31
The ionized guanidino group has a planar structure and is largely unreactive due to the resonance within the guanidino group, but can be activated if an intramolecular proton acceptor is available in the neighborhood. Under acidic conditions, the guanidino group reacts in the protonated form, while at neutral – alkaline pH it reacts in the unprotonated form.32 In this ionic environment, Arg with the covalently linked CHD is a strong electrophile,33 allowing to react with its vacant orbitals to an electron-rich centre, like with acidic amino acids. It is expected that the “covalent”-like modification will result in a reduced hydration at the terminal guanidino NH2 groups, leading to a decreased screening of the charges of the polyP phosphate groups by the solvent. As a consequence, the electrostatic interaction between Arg and phosphate will increase.
We show that after cyclohexanedione derivatization and stabilization in the presence of borate, the inhibitory sensitivity of RBD increases. The experiments revealed that the RBD, carrying the DHCH-Arg-borate complex, is much more susceptible to polyP inhibition than the unmodified RBD. More recent calculations revealed that Arg side chains are always charged, even at pH values around 10. This finding reveals that at the high pKa value of the Arg guanidinium group (13.8), these side chains are invariably protonated under physiological conditions.34 This polarization state is valid even if the guanidinium moiety is buried in a hydrophobic microenvironment.
Results
Surface functional patch prediction
A rational approach to design an activity modulator of a peptide within a metabolic net of an organism has to implement surface model prediction. These structure-based functional calculations highlight the distribution/pattern of aa with related physicochemical properties, followed by hydrophobicity/hydrophilicity predictions, and also include the charge-density distributions which will help to design potential binding ligands. These data are especially important for the RBD of the SARS-CoV-2 S-protein since this domain is not enzymatically active, but prone to ligand-caused conformational changes.35Alignment of polyP on the RBD
The association of the polyP chain to the RBD was predicted again by applying the Chimera program. The overall location of the polyP chain (a sequence of 15 Pi units has been chosen) along the polyP groove is shown in Fig. 2A. The rationale for using the 15 units long polyP for the in silico study was the finding (W. E. G. Müller; unpublished) that the polyP40 sample which was used for the in vitro inhibition experiments will rapidly undergo an enzymatic hydrolysis to a polyP∼15 long polymer in vivo.41 In the model presented here, the binding of polyP15 to the surface of the RBD of the S-protein is depicted. There, the polyP chain spans about half of the RBD in length. A closer view of the calculated extension highlights the association of the polyP chain to the Arg/Lys residues of the RBD at positions 457, 462, 466, 355 and 357 (Fig. 2B). Due to the high flexibility of the polymer,14 the association of the polyP chain with the surface of the RBD is tight and spans a distance between 2.5 and 3.3 Å between the attachment sites of the polymer and the Arg residues, matching also with the published distances between Arg and phosphate;42Fig. 2C. Four Arg residues at positions 457, 466, 355 and 357 interact with the polyP chain. The four attachment sites within the polyP chain and RBD have a spacing of 5 Pi units at one end (between Arg-457 and Arg-466) and 3 Pi units in the middle and again 5 Pi units at the terminus (Arg-355 and Arg-357). The uneven number of the Pi units within the polyP chain reveal a staggered configuration of the polymer between these attachment points. It has been pointed out that polyP has a high degree of rotational flexibility, paralleled with various conformations, like staggered or eclipsed, which depend on the arrangement of the tetrahedral PO4 units.13,43 | ||
| Fig. 2 Association of a polyP chain with the surface of the RBD within the S-protein. (A) Overall view of the model of the RBD, associated with polyP. The surface of the RBD is shaded. (B) The attachment site on the surface of RBD, presented by space-filling models of the aa, is given and the Arg residues 457, 466, 355 and 357 are marked (colored in blue). The two aa (only Asp [D] is shown), which are interspersing the Arg cluster, are colored in red. (C) The three-dimensional molecular model of the attachment site of the Arg residues with the polyP chain is shown; the distances between this amino acid (at positions 457, 466, 355 and 357) and the four Pi units within the polyP chain are given; they measure between 2.6 and 3.4 Å. | ||
Modification of Arg with 1,2-cyclohexanedione
The two basic aa Lys and Arg have high aqueous pKa values [12–13.7 for Arg, and ∼10.5 for Lys], which in turn allow them to carry at physiological pH a considerable charge that gives rise to strong electrostatic interactions. Consequently, at those patches at the biomolecular surfaces a strong interaction with a first water layer through electric forces as well as local H bonds can be set up, which allow the spatial arrangement of water molecules that can grow to up to a bulky, 1–1.5 nm thick hydration shell.44 The surrounding water molecules form a gradient of dipole dynamics, which is correlated with an increase in ligand reactivity.45 The modification of Arg with 1,2-cyclohexanedione (CHD), forming DHCH-arginine (dihydroxycyclohex-2-ylene adduct of Arg), results in a bending of the guanidino group out of the overall surface plane (Fig. 3). Since the hydration layer is depleted around the more extended side chain (around the guanidino group) of Arg, an increased reactivity of the residue with incoming molecules can be deduced (Fig. 3).46 In turn, we applied both the unmodified versus the modified RBD for the following inhibition studies.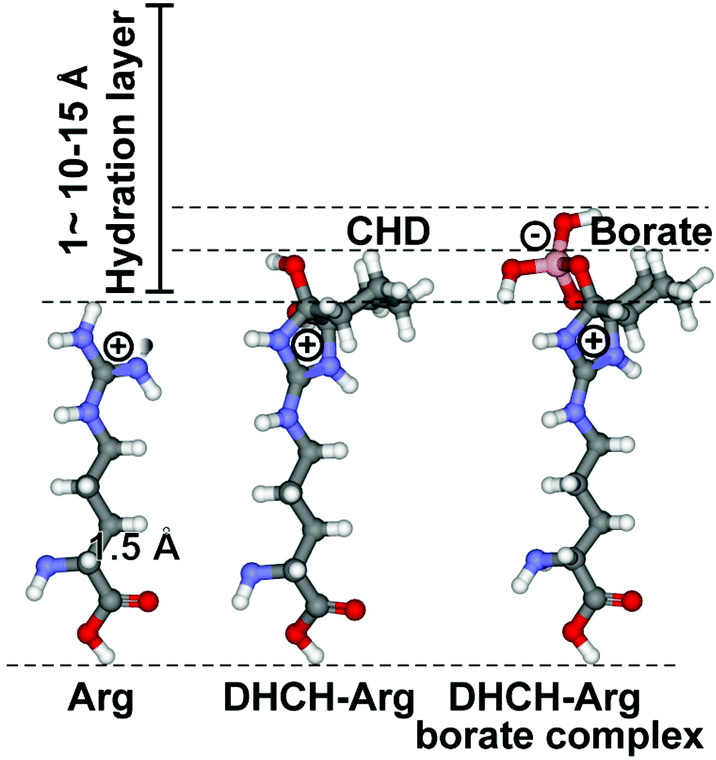 | ||
| Fig. 3 Increased reactivity of the guanidino terminus of Arg after the selective modification of this residue with CHD. At pH 9 (in borate buffer), DHCH-Arg is formed, which is stabilized by complex formation with borate. In comparison with the length of the C–C single bond (1.54 Å), the hydration layer around a molecule, which measures 10–15 Å, is bulky. The increased polarization of the electron shell around the guanidino terminus of Arg has been attributed to the enhanced polarity of DHCH-Arg. | ||
Differential inhibition of RBD by unmodified/modified Arg
PolyP, in the form of Na-polyP with a chain length of ∼40 Pi units, significantly inhibits the binding of the RBD to the cellular ACE2 immobilized at the bottom of the well. Addition of 1 μg mL−1 of Na-polyP to the system results in an inhibition by 20%. At concentrations higher than 1 μg mL−1, values of ∼50% are reached (Fig. 4A). After the selective modification of the Arg residues in the RBD viral binding protein with CHD, dissolved in the borate buffer, to DHCH-Arg, a substantial increase in the inhibitory activity of polyP in the RBD:ACE2 binding assay is observed. At 1 μg mL−1, a large inhibition of the system by 79% is recorded. Interesting is the fact that also at 0.1 μg mL−1 of Na-polyP, the inhibition measures 59%; under these concentration conditions, no significant reduction of the binding of Na-polyP to the unmodified RBD is measured (not shown here). Increasing the concentration to 10 μg mL−1 and to 30 μg mL−1 in the system with modified Arg residues in the RBD further increases the inhibitory effect of polyP (Fig. 4A).
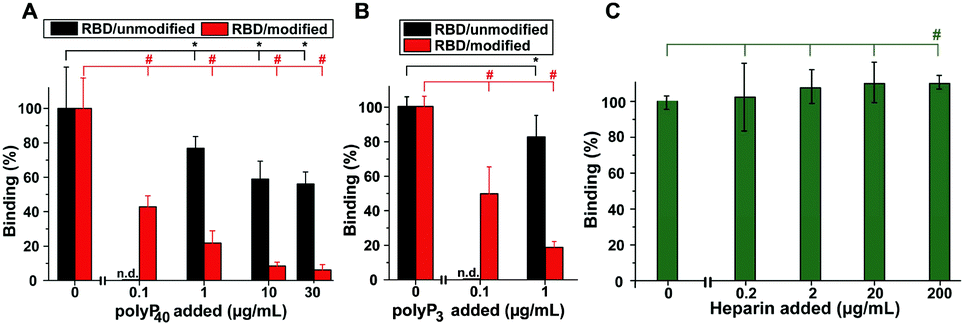 | ||
| Fig. 4 Effect of polyP and heparin on the binding of the RBD to ACE2. (A) Influence of polyP (polyP40) on the binding reaction containing either the unmodified RBD peptide (black bars) or the CHD-modified (DHCH-Arg) RBD (red bars). (B) Effect of polyP3 on the RBD:ACE2 interaction with the unmodified RBD (black) or the DHCH-Arg modified RBD. (C) Enhancement of the RBD:ACE2 binding by heparin. | ||
Also the Na-polyP trimer (polyP3) at concentrations ≥1 μg mL−1 significantly inhibits RBD:ACE2 binding, if in the RBD the Arg residues are not modified to DHCH-Arg (Fig. 4B). The highest inhibition is reached with 50% only at concentrations >30 μg mL−1 (not shown). In the system with the Arg-modified RBD, a much stronger reduction of the binding affinity is observed with 80% at 1 μg mL−1 and still with ∼50% at the low concentration of 0.1 μg mL−1. The corresponding IC50 values for the RBD:ACE2 binding inhibition are 43 ± 7 μg polyP per mL (non-modified Arg in the RBD) and 0.08 ± 0.4 μg polyP per mL (DHCH-Arg modified).
Simultaneously, we substituted the 10 mM HEPES buffer in the binding assay with Hanks’ balanced salt solution (#55021C SAFC; Sigma) composed of a physiological composition of ions. Addition of polyP to this system did not significantly affect the inhibitory activity of polyP (data not shown).
Enhancement of RBD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :ACE2 binding by heparin
:ACE2 binding by heparin
In a comparative series of experiments, the effect of heparin, suspected to interfere with the RBD:ACE2 binding by affecting the RBD at the Arg residues under conformational changes, was studied. Heparin was tested in the same RBD:ACE2 binding assay. Surprisingly, this anticoagulant causes no significant effect on the interaction between RBD and ACE2 at lower concentrations (<20 μg mL−1). Only at the high concentration of 200 μg mL−1, a small (by 12%) but significant increase of the binding could be determined.
Discussion
Based on epidemiological studies, it is obvious that the status of innate immunity contributes to the manifestation of the disease. On the cellular level, the B lymphocytes as well as the T lymphocytes primarily execute the adaptive branch of the immune system, while the large granular lymphocytes/natural killer (NK) cells are critical for the innate immune defense. The NK cells kill the viral-infected cells and protect the organisms against viral infection and disease (reviewed in: ref. 47). These cells secrete interferons and cytokines, IFNγ and TNFα, which act on macrophages or dendritic cells and enhance immune response. The number of NK cells and also one of the platelets, a further arm of innate immunity, decrease during the progression of the corona disease.48 Lymphocytes and especially the platelets are rich in polyP,10,49 the proposed relevant inorganic polymer involved. It has to be stressed here that polyP is released from the platelets in two forms, as <100 Pi units long free and short polymer and as nanoparticles (∼200 nm) containing a long-chain polyP.50,51 Only for the long-chain polyP polymers, a modulating effect on blood coagulation and innate immunity has been reported.7,52 Results on the role of short-chain polyP have been published for the first time only recently.53 In this report, it has been shown that polyP acts on both macrophage and fibrocyte differentiation.Some trace elements are known to bind to virions and control their pathogenicity, like the NS3/4A protein, a membrane-targeting serine protease of the hepatitis C virus. This NS3-4A serine protease binds to zinc resulting in a subsequent conformation change. These trace elements are also linked to the oxidant/antioxidant balance system of the host cells, a status of the cells which in turn circularly commands the infectivity of the virus.54 In contrast to trace elements, the cellular inorganic polymer polyP studied here binds to the RBD of the SARS-CoV-2 S-protein and causes an inactivation of the viral function on the level of attachment of the virus to the cellular membrane, the specific ACE2 receptor.
In the present study, polyP preparation with a chain length of 40 Pi units has been selected to assess the inhibition potential of inorganic polyP on the interaction between the COVID-19 spike RBD and the cellular receptor ACE2. A 50% reduction of the binding affinity was measured at concentrations of ≥10 μg mL−1. The binding of polyP chains has been pinpointed to the Arg residues 457, 466, 355 and 357, most likely flanked by the likewise basic aa, Lys at positions 458 and 356. Interestingly enough, the number and arrangement of the Arg residues within the RBD of the SARS-CoV-2 S-protein are highly conserved; to date, all the sequences listed in the database NCBI_Blast comprise the Arg moieties at the same position.
The charged aa at the surface of the RBD form a groove of hydrophilic aa with a hydrophobicity index of −4.5 (Fig. 1A and B). These calculations are also supported by the determination of the electric charge distribution on the surface of the RBD which again revealed a hydrophilic furrow with a value of ∼+2 (Fig. 1C). The main attachment sites of polyP have been determined to occur at the Arg residues 457, 466, 355 and 357, allowing the binding of polyP stretches with five Pi at the termini of the chain and the central polyP3 part (Fig. 2).
After the chemical modification of the Arg residues within the RBD with CHD to DHCH-Arg, the modified RBD becomes highly sensitive towards polyP. During this transformation of Arg to DHCH-Arg, most significantly at the Arg position 457/466, which is traversed by the two acidic aa Asp (467) and Glu (465), an effective intramolecular proton acceptor interaction can occur36 and an ionic salt bridge bond can be formed.55 From this center, additional electrostatic interactions originate,27 which have the potential to interact with polyP. Based on a molecular dynamics study, it has been proposed that hydration of a bidentate complex of the Arg guanidino group significantly increases the polarity of the N–H bonds with a subsequently increased positive charge at each N–H hydrogen which can interact with the oxygen in water.56 Additionally, the modification to DHCH-Arg allows a protrusion of the surface Arg residues to the surrounding hydration shell, which is paralleled with an increase of the polarization of the electron shell around the guanidino terminus of DHCH-Arg. This interplay might facilitate the accessibility of the RBD for polyP. As a consequence, after the transformation of Arg to DHCH-Arg, the RBD:ACE2 binding system is inhibited by polyP concentrations as low as 0.1 μg mL−1. Interestingly, this concentration is even lower than the one which exists in the circulating blood with 1 to 3 μg mL−1, after full platelet activation (reviewed in: ref. 49). The half-life of polyP in human blood or plasma is ∼1.5 to 2 h.57 The binding of polyP to the RBD has been found to be a stoichiometric one. Calculating on the basis of the molecular weight of the RBD [∼24500 Da], added at a concentration of 2 μg mL−1, and the concentration of Na-polyP with 0.1 μg ml−1 (the concentration of polyP40 causing a 50% inhibition of the system), the molar ratio between these two components is calculated. This ratio of 8.47 × 10−8 mmol (for a polyP chain with an average chain length of ∼15 Pi units) to 8.16 × 10−8 mmol (RBD) is close to 1:1, the theoretical stoichiometric ratio between the two components. For this calculation, a polyP with a chain length of 15 Pi units was used in order to compensate for a potential reduction of the chain length by the action of the alkaline phosphatase, the polyP hydrolyzing enzyme.41,58 A comparable stoichiometry for the binding of the shorter polyP, the polyP3, to the RBD has been calculated; again a ratio of about 1:1 is found. The modeling approach supports this inhibition and is in agreement with the binding of polyP3 to the Arg residues 466 and 355, leaving space for such a polymer length.
A quantitative analysis of the physiological relevance of the binding assay, used in the present study, revealed that the potency of the antiviral effect of polyP matches realistic, pathophysiological circumstances. It has been analyzed by qRT-PCR that the mean log10 viral load in COVID-19 patients amounts to 5.6 copies per mL.59 In our binding assay used, 10 ng of a RBD preparation (size of 23.6 kDa) have been added to the test system. Based on this value, a concentration of RBD of 2.5 × 1011 molecules per 100 μL (2.5 × 1012 molecules per mL) is present in the system. Considering that the spike protein is a trimer60 and ∼40 spikes are present on the surface of each virus particle (the total number of RBD per virus particle: 120),61 the binding assay contains about the same RBD concentration compared to the in vivo load with the RBD on the virus particles.
In a recent study, it has been proposed that the glycan coating around the SARS-CoV-2 S protein could reduce the therapeutic efficacy of potential drugs.62 However, the available modeling study disclosed that only two glycans can be expected to exist at that RBD which is suspected to be the binding site for polyP.60 These sugar units do not shield the region of interaction of polyP with the RBD.
Considering the published data18 showing that heparin interacts with the RBD and causes a conformational change, a reduction of the binding affinity between the RBD and ACE2 could be assumed. However, instead, our presented data revealed an increase in the binding strength at a stoichiometric ratio of 1:5 (heparin:RBD). This finding might suggest that the RBD undergoes conformational changes that result in a cooperative interaction on the binding between RBD and ACE2, similar to the one determined for the RBD during the entry phase of the virus into the host cell.63 Taken together, the presented results demonstrate that the physiological polymer polyP inhibits the binding of the SARS-CoV-2 binding domain, RBD, to the host cell ACE2 receptor. These data have been obtained with the recombinant binding partner in a solid phase ligand/receptor system. Experiments are now in progress using intact cells that express the ACE2 receptor together with anti-ACE2 antibodies and the biotin-labeled viral RBD to investigate more realistically, but simultaneously in a defined test setup, the effect of polyP on the initial viral-cell attachment step.
Conclusion
In the present report, it is demonstrated that polyP, with the characteristics which the polymer has in the circulating blood, abolishes the early phase of the SARS-CoV-2 binding to the target cell via the ACE2 receptor. To highlight, at the concentrations of polyP used (<10 μg mL−1) and with polyP of a chain length of <100 Pi units, no effect of the polymer on blood clotting has been reported.51,64 Based on the data presented here, we propose to use polyP as the biologically active ingredient in a novel anti-viral/COVID-19 drug, applied in nasal and pharyngeal sprays for, in the beginning, clinical studies.Materials and methods
Materials
Sodium tripolyphosphate [3 phosphate (Pi) units; polyP3] was purchased from Sigma (#238503; Taufkirchen; Germany). Na-polyphosphate (Na-polyP) with an average chain length of 40 Pi units (Na-polyP40) was from Chemische Fabrik Budenheim (Budenheim; Germany) and heparin [sodium salt from porcine intestinal mucosa; ≥180 USP units per mg; molecular weight 11349 g mol−1 (ref. 65)] from Sigma (#H3393; Taufkirchen; Germany).
Prediction/mapping of the peptide surfaces of the RBD
Binding assay for the RBD of S-protein to the cellular ACE2
The sensitivity of the binding between the RBD and the cellular ACE2 was determined using the RBD (SARS-CoV-2):ACE2 Inhibitor Screening Assay Kit (BPS Bioscience/Tebu-bio, Offenbach; Germany). In this binding assay, the recombinant ACE2 (50 ng per well) was bound to the bottom of the 96-well plate, while the RBD of the S1-protein (100 ng per well) was labeled with biotin. The complex RBD:ACE2 was quantitated with streptavidin-HRP (horseradish peroxidase) after addition of the HRP substrate. PolyP was added to the RBD for 60 min (23 °C) and subsequently exposed to ACE2. The chemiluminescence produced was quantitated using a PerkinElmer-Wallac victor 3 V multi-label microplate reader (PerkinElmer, Waltham, MA; USA). From the resulting measured readings, the blank (composed of the immune buffers and the loosely bound binding components) was subtracted. The values obtained for the binding of the RBD to ACE2 in the absence of the inhibitor were referred to as positive controls and set to 100%. The non-modified RBD was added in 10 mM HEPES (pH 7.0) buffer to the system. In parallel, the CHD-arginine modified RBD was added to the assay in the 20 mM Na-borate buffer (pH 9).
Modification of Arg
A selective modification of the Arg residues in the RBD was performed with 1,2-cyclohexanedione [CHD] (#W345806; Sigma) in a 20 mM Na-borate buffer (pH 9).31 The reaction was run at 23 °C for 2 h under the formation of DHCH-arginine (N7,N8-1,2-dihydroxycyclohex-1,2-ylene-L-arginine), which forms a complex with borate. Therefore, the substrate RBD (2 pmoI mL−1) was treated with 0.15 M CHD in 0.25 M Na-borate buffer at pH 9.0 for 2 h.31Statistical analysis
From the experimental results, the average ± standard deviations (σ) were determined. Student's t-test was applied to perform comparisons between the two groups. The average values and σ originated from at least three independent experiments. The RBD:ACE2 binding assay is highly sensitive. Therefore, less frequently, results from different series of experiments have been pooled. Values of p < 0.05 were considered statistically significant (*; #). The calculations were performed with the GraphPad Prism 7.0 software (GraphPad Software, La Jolla; CA). IC50 values (half-maximal inhibitory concentration) were calculated as described.68
Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. E. G. M. is the holder of an ERC Advanced Investigator Grant (no. 268476). In addition, W. E. G. M. has obtained three ERC-PoC grants (Si-Bone, no. 324564; MorphoVES-PoC, no. 662486; and ArthroDUR, no. 767234). Further support came from the BMBF (Chinese–German cooperation in industry-led collaborative projects; “SKIN-ENERGY” – FKZ 13GW0403B). Finally, this work was supported by grants from the European Commission (grant numbers 604036 and 311848), the BiomaTiCS research initiative of the University Medical Center, Mainz, and from BMWi-ZIM (ZF4294002 AP9).References
- W. E. G. Müller, B. Blumbach and I. M. Müller, Transplantation, 1999, 68, 1215 CrossRef.
- S. Tripathi, M. R. White and K. L. Hartshorn, Innate Immun., 2015, 21, 73 CrossRef.
- S. Akira, S. Uematsu and O. Takeuchi, Cell, 2006, 124, 783 CrossRef CAS.
- F. Roth-Walter, A. B. Riemer, E. Jensen-Jarolim and H. Stockinger, in Comparative Medicine, ed. E. Jensen-Jarolim, Springer-Press, Wien, 2014, pp. 219–266 Search PubMed.
- A. Mantovani and C. Garlanda, Nat. Immunol., 2013, 14, 768 CrossRef CAS.
- C. H. Wong, C. N. Jenne, B. Petri, N. L. Chrobok and P. Kubes, Nat. Immunol., 2013, 14, 785 CrossRef CAS.
- J. M. Wat, J. H. Foley, M. J. Krisinger, O. L. Mae, V. Lei, G. A. Wasney, E. Lameignere, N. C. Strynadka, S. A. Smith, J. H. Morrissey and E. M. Conway, Blood, 2014, 123, 768 CrossRef CAS.
- S. Khandelwal and G. M. Arepally, Thromb. Haemostasis, 2016, 116, 792 CrossRef.
- S. H. Yun, E. H. Sim, R. Y. Goh, J. I. Park and J. Y. Han, BioMed. Res. Int., 2016, 2016, 9060143 Search PubMed.
- B. Lorenz, J. Leuck, D. Köhl, W. E. G. Müller and H. C. Schröder, J. Acquired Immune Defic. Syndr. Hum. Retrovirol., 1997, 14, 110 CrossRef CAS.
- W. E. G. Müller, K. Renneisen, M. H. Kreuter, H. C. Schröder and I. Winkler, J. Acquired Immune Defic. Syndr., 1988, 1, 453 Search PubMed.
- E. Keyaerts, L. Vijgen, C. Pannecouque, E. J. M. Van Damme, W. J. Peumans, H. Egberink, J. Balzarini and M. Van Ranst, Antiviral Res., 2007, 75, 179 CrossRef CAS.
- W. E. G. Müller, H. C. Schröder and X. H. Wang, Chem. Rev., 2019, 119, 12337 CrossRef.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N. H. Wu, A. Nitsche, M. A. Müller, C. Drosten and S. Pöhlmann, Cell, 2020, 181, 271 CrossRef CAS.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang, L. Zhang and X. Wang, Nature, 2020, 581, 215 CrossRef CAS.
- X. Ou, Y. Liu, X. Lei, P. Li, D. Mi, L. Ren, L. Guo, R. Guo, T. Chen, J. Hu, Z. Xiang, Z. Mu, X. Chen, J. Chen, K. Hu, Q. Jin, J. Wang and Z. Qian, Nat. Commun., 2020, 11, 1620 CrossRef CAS.
- E. Vicenzi, F. Canducci, D. Pinna, N. Mancini, S. Carletti, A. Lazzarin, C. Bordignon, G. Poli and M. Clementi, Emerging Infect. Dis., 2004, 10, 413 CrossRef CAS.
- C. Mycroft-West, D. Su, S. Elli, S. Guimond, G. Miller, J. Turnbull, E. Yates, M. Guerrini, D. Fernig, M. Lima and M. Skidmore, bioRxiv, 2020 DOI:10.1101/2020.02.29.971093.
- G. Choijilsuren, R. S. Jhou, S. F. Chou, C. J. Chang, H. I. Yang, Y. Y. Chen, W.-L. Chuang, M.-L. Yu and C. Shih, Sci. Rep., 2017, 7, 14461 CrossRef.
- C. J. Mycroft-West, D. Su, I. Pagani, T. R. Rudd, S. Elli, S. E. Guimond, G. Miller, M. C. Z. Meneghetti, H. B. Nader, Y. Li, Q. M. Nunes, P. Procter, N. Mancini, M. Clementi, N. R. Forsyth, J. E. Turnbull, M. Guerrini, D. G. Fernig, E. Vicenzi, E. A. Yates, M. A. Lima and M. A. Skidmore, bioRxiv, 2020 DOI:10.1101/2020.04.28.066761.
- C. Mycroft-West, D. Su, S. Elli, S. Guimond, G. Miller, J. Turnbull, E. Yates, M. Guerrini, D. Fernig, M. Lima and M. Skidmore, bioRxiv, 2020 DOI:10.1101/2020.02.29.971093.
- S. Y. Kim, W. Jin, A. Sood, D. W. Montgomery, O. C. Grant, M. M. Fuster, L. Fu, J. S. Dordick, R. J. Woods, F. Zhang and R. J. Linhardt, Antiviral Res., 2020, 181, 104873 CrossRef CAS.
- W. E. G. Müller, T. Okamoto, P. Reuter, D. Ugarkovic and H. C. Schröder, J. Biol. Chem., 1990, 265, 3803 Search PubMed.
- G. Gasparini, E. K. Bang, J. Montenegro and S. Matile, Chem. Commun., 2015, 51, 10389 RSC.
- C. J. Chen, K. C. Tsai, P. H. Kuo, P. L. Chang, W. C. Wang, Y. J. Chuang and C. M. Dah-Tsyr, BioMed. Res. Int., 2015, 2015, 237969 Search PubMed.
- N. Sakai, N. Sorde, G. Das, P. Perrottet, D. Gerard and S. Matile, Org. Biomol. Chem., 2003, 1, 1226 RSC.
- A. S. Woods and S. Ferré, J. Proteome Res., 2005, 4, 1397 CrossRef CAS.
- B. Westenberg, T. Boller and A. Wiemken, FEBS Lett., 1989, 254, 133 CrossRef CAS.
- M. A. Tortorici, A. C. Walls, Y. Lang, C. Wang, Z. Li, D. Koerhuis, G. J. Boons, B. J. Bosch, F. A. Rey, R. J. de Groot and D. Veesler, Nat. Struct. Mol. Biol., 2019, 26, 481 CrossRef.
- N. Ngernyuang, W. Yan, L. M. Schwartz, D. Oh, Y. B. Liu, H. Chen and R. Shao, Neoplasia, 2018, 20, 182 CrossRef CAS.
- L. Patthy and E. L. Smith, J. Biol. Chem., 1975, 250, 557 CAS.
- L. Patthy and J. Thész, Eur. J. Biochem., 1980, 105(2), 387 CrossRef CAS.
- L. R. Domingo, M. J. Aurell, P. Perez and A. Saeza, RSC Adv., 2012, 2, 1334 RSC.
- C. A. Fitch, G. Platzer, M. Okon, B. E. Garcia-Moreno and L. P. McIntosh, Protein Sci., 2015, 24, 752–761 CrossRef CAS.
- R. Yan, Y. Zhang, Y. Li, L. Xia, Y. Guo and Q. Zhou, Science, 2020, 367, 1444 CrossRef CAS.
- D. Suckau, M. Mak and M. Przybylski, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5630 CrossRef CAS.
- L. Li, I. Vorobyov and T. W. Allen, J. Phys. Chem. B, 2013, 117, 11906 CrossRef CAS.
- J. R. Fromm, R. E. Hileman, E. E. Caldwell, J. M. Weiler and R. J. Linhardt, Arch. Biochem. Biophys., 1995, 323, 279 CrossRef CAS.
- L. Lorenzo-Orts, U. Hohmann, J. Zhu and M. Hothorn, Life Sci. Alliance, 2019, 2, e201900385 CrossRef.
- J. Kyte and R. F. Doolittle, J. Mol. Biol., 1982, 157, 105 CrossRef CAS.
- B. Lorenz and H. C. Schröder, Biochim. Biophys. Acta, 2001, 1547, 254 CrossRef CAS.
- M. J. Harms, J. L. Schlessman, G. R. Sue and B. García-Moreno, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18954 CrossRef CAS.
- J. Majling and F. Hanic, Top. Phosphorus Chem., 1980, 10, 341 CAS.
- D. Laage, T. Elsaesser and J. T. Hynes, Chem. Rev., 2017, 117, 10694 CrossRef CAS.
- M. Maurer and C. Oostenbrink, J. Mol. Recognit., 2019, 32, e2810 CrossRef CAS.
- D. Shah and A. R. Shaikh, J. Biomol. Struct. Dyn., 2016, 34, 104 CrossRef CAS.
- M. Market, L. Angka, A. B. Martel, D. Bastin, O. Olanubi, G. Tennakoon, D. M. Boucher, J. Ng, M. Ardolino and R. C. Auer, Front. Immunol., 2020, 11, 1512 CrossRef CAS.
- C. Qin, L. Zhou, Z. Hu, S. Zhang, S. Yang, Y. Tao, C. Xie, K. Ma, K. Shang, W. Wang and D. S. Tian, Clin. Infect. Dis., 2020, ciaa248, DOI:10.1093/cid/ciaa248.
- J. H. Morrissey, S. H. Choi and S. A. Smith, Blood, 2012, 119, 5972 CrossRef CAS.
- J. J. Verhoef, A. D. Barendrecht, K. F. Nickel, K. Dijkxhoorn, E. Kenne, L. Labberton, O. J. T. McCarty, R. Schiffelers, H. F. Heijnen, A. P. Hendrickx, H. Schellekens, M. H. Fens, S. de Maat, T. Renné and C. Maas, Blood, 2017, 129, 1707 CrossRef CAS.
- J. I. Weitz and J. C. Fredenburgh, Blood, 2017, 129, 1574 CrossRef CAS.
- E. M. Conway, Front. Med., 2019, 6, 67 CrossRef.
- P. M. Suess, L. E. Chinea, D. Pilling and R. H. Gomer, J. Immunol., 2019, 203, 493 CrossRef CAS.
- U. C. Chaturvedi, R. Shrivastava and R. K. Upreti, Curr. Sci., 2004, 87, 1536 CAS.
- T. N. Bat, G. A. Bentley, T. O. Fischmann, G. Boulot and R. J. Poljak, Nature, 1990, 347, 483 CrossRef.
- D. Frigyes, F. Alber, S. Pongor and P. Carloni, J. Mol. Struct., 2001, 574, 39 CrossRef CAS.
- S. A. Smith, N. J. Mutch, D. Baskar, P. Rohloff, R. Docampo and J. H. Morrissey, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 903 CrossRef CAS.
- S. Omelon, J. Georgiou, Z. J. Henneman, L. M. Wise, B. Sukhu, T. Hunt, C. Wynnyckyj, D. Holmyard, R. Bielecki and M. D. Grynpas, PLoS One, 2009, 4, e5634 CrossRef.
- E. Pujadas, F. Chaudhry, R. McBride, F. Richter, S. Zhao, A. Wajnberg, G. Nadkarni, B. S. Glicksberg, J. Houldsworth and C. Cordon-Cardo, Lancet Respir. Med., 2020, 8(9), e70 CrossRef CAS.
- A. C. Walls, Y. J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Cell, 2020, 181, 281 CrossRef CAS.
- B. Turoňová, M. Sikora, C. Schürmann, W. J. H. Hagen, S. Welsch, F. E. C. Blanc, S. von Bülow, M. Gecht, K. Bagola, C. Hörner, G. van Zandbergen, S. Mosalaganti, A. Schwarz, R. Covino, M. D. Mühlebach, G. Hummer, J. K. Locker and M. Beck, bioRxiv, 2020 DOI:10.1101/2020.06.26.173476.
- L. Casalino, Z. Gaieb, A. C. Dommer, A. M. Harbison, C. A. Fogarty, E. P. Barros, B. C. Taylor, E. Fadda and R. E. Amaro, bioRxiv, 2020 DOI:10.1101/2020.06.11.146522.
- J. Shang, Y. Wan, C. Luo, G. Ye, Q. Geng, A. Auerbach and F. Li, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 11727 CrossRef CAS.
- T. L. Lindahl, S. Ramström, N. Boknäs and L. Faxälv, Biochem. Soc. Trans., 2016, 44, 35 CrossRef CAS.
- PubChem Database, National Center for Biotechnology Information, Heparin, CID = 772, https://pubchem.ncbi.nlm.nih.gov/compound/Heparin (accessed July 2020).
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605 CrossRef CAS.
- H. A. Basta, S. Ashraf, J. Y. Sgro, Y. A. Bochkov, J. E. Gern and A. C. Palmenberg, Virology, 2014, 448, 82 CrossRef CAS.
- S. Aykul and E. Martinez-Hackert, Anal. Biochem., 2016, 508, 97 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |