
The unique potency of Cowpea mosaic virus (CPMV) in situ cancer vaccine†

Abstract
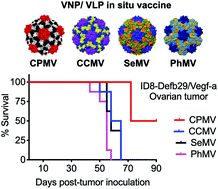
The immunosuppressive tumor microenvironment enables cancer to resist immunotherapies. We have established that intratumoral administration of plant-derived Cowpea mosaic virus (CPMV) nanoparticles as an in situ vaccine overcomes the local immunosuppression and stimulates a potent anti-tumor response in several mouse cancer models and canine patients. CPMV does not infect mammalian cells but acts as a danger signal that leads to the recruitment and activation of innate and subsequently, adaptive immune cells. In the present study we addressed whether other icosahedral viruses or virus-like particles (VLPs) of plant, bacteriophage and mammalian origin can be similarly employed as intratumoral immunotherapy. Our results indicate that CPMV in situ vaccine outperforms Cowpea chlorotic mottle virus (CCMV), Physalis mosaic virus (PhMV), Sesbania mosaic virus (SeMV), bacteriophage Qβ VLPs, or Hepatitis B virus capsids (HBVc). Furthermore, ex vivo and in vitro assays reveal unique features of CPMV that makes it an inherently stronger immune stimulant.
- This article is part of the themed collection: Biomaterials Science Most Popular 2020
 Please wait while we load your content...
Please wait while we load your content...

