
Biomaterial-based approaches to engineering immune tolerance
Amy E.
Emerson†
,
Emily M.
Slaby†
,
Shivani C.
Hiremath†
and
Jessica D.
Weaver
 *
*
School of Biological and Health Systems Engineering, Arizona State University, Tempe, AZ, USA. E-mail: j.weaver@asu.edu
First published on 5th November 2020
Abstract
The development of biomaterial-based therapeutics to induce immune tolerance holds great promise for the treatment of autoimmune diseases, allergy, and graft rejection in transplantation. Historical approaches to treat these immunological challenges have primarily relied on systemic delivery of broadly-acting immunosuppressive agents that confer undesirable, off-target effects. The evolution and expansion of biomaterial platforms has proven to be a powerful tool in engineering immunotherapeutics and enabled a great diversity of novel and targeted approaches in engineering immune tolerance, with the potential to eliminate side effects associated with systemic, non-specific immunosuppressive approaches. In this review, we summarize the technological advances within three broad biomaterials-based strategies to engineering immune tolerance: nonspecific tolerogenic agent delivery, antigen-specific tolerogenic therapy, and the emergent area of tolerogenic cell therapy.
 Amy E. Emerson | Amy Emerson is a Ph.D. student in the School of Biological and Health Systems Engineering at Arizona State University. Her current research interests include the development of translatable, biomaterial-based technologies for cell-based therapies and Type 1 Diabetes. |
 Emily M. Slaby | Emily Slaby is a Ph.D. student in the School of Biological and Health Systems Engineering at Arizona State University. Her current research focuses on the development of synthetic hydrogel platforms to support tolerogenic cells for transplant applications. In general, she is interested in engineering cell therapies to treat autoimmune diseases and cancer. |
 Shivani C. Hiremath | Shivani Hiremath is an international student from India currently pursuing a master's degree in biomedical engineering at the School of Biological and Health Systems Engineering at Arizona State University. Her work at the Weaver Lab at Arizona State University centers around immune engineering and biomaterial science. |
 Jessica D. Weaver | Dr Jessica D. Weaver earned her PhD in Biomedical Engineering at the University of Miami. She completed a postdoctoral fellowship at the Georgia Institute of Technology, where she was supported by the NIH ILET2 training grant and a JDRF Postdoctoral Fellowship. As an Assistant Professor in the School of Biological and Health Systems Engineering at Arizona State University, Dr Weaver's research centers on developing translatable cell-based therapies for the treatment of disease, with a focus on islet transplantation for the treatment of Type 1 Diabetes. The Weaver lab uses biomaterials and tolerogenic immunoengineering approaches with the aim to generate immunosuppression-free transplantation strategies. |
1. Introduction
Immunological tolerance was described by Peter Medawar in 1961 as “a state of indifference or non-reactivity” toward a typically immunogenic substance.1 The term was originally used in the context of transplantation immunity, where it was observed that foetal or new-born mice accepted grafts from immunologically discordant mouse strains, thereby “actively acquiring” tolerance toward grafts of those tissues that persisted into adulthood.2 In the 60 years since Medawar's landmark publication and consequent Nobel Prize, extensive work has been devoted to elucidating and understanding the mechanisms of immune tolerance.3 Concurrently, the evolution of the field of biomaterials has led to a greater understanding of how materials interact with cells of the immune system,4 enabling the convergence of these two fields for the development of biomaterials-based tolerogenic therapies.While tolerance was first coined in the context of allogeneic transplantation, it is now recognized that defects in the mechanisms of tolerance result in the development of allergies and autoimmune diseases.5 Over 80 distinct autoimmune diseases have been identified that impact up to 8% of the population in the United States.6 Normal physiological tolerance is maintained through the mechanisms of (1) central tolerance, where the immune system is instructed in discriminating self from non-self within the thymus during T cell development and the bone marrow during B cell development;7 and (2) peripheral tolerance, maintained in secondary lymphatic organs via autoreactive T cell clonal deletion, induction of T cell anergy, or conversion of T cells to immunosuppressive regulatory T cells (Tregs) in response to antigen presentation by innate immune cells such as macrophages and dendritic cells.8 Allergies result from defects in peripheral tolerance mechanisms, whereas autoimmune diseases are a consequence of dysfunction in both central and peripheral tolerance.
Much of the precise mechanisms of establishing peripheral tolerance remain to be fully elucidated, which makes tolerogenic therapeutic development challenging.9 In central tolerance development, T cells undergo positive selection in the thymus, resulting in a T cell receptor repertoire that is autoreactive. Approximately 50% of positively selected autoreactive T cells undergo thymic deletion, known as negative selection, and the remainder mature and enter the periphery.10 To maintain peripheral T cell tolerance of self–antigens, a variety of cells and molecules must act through multiple mechanisms. Some circulating autoreactive T cells undergo further deletion within the lymph nodes,11 and dysfunction in this process may contribute to autoimmunity.12 Currently, it is unclear whether peripheral autoreactive T cells must be continuously suppressed to maintain tolerance or if they require antigen presentation under the correct circumstances.13
Biomaterials are an integral tool in the development of delivery vehicles in immunotherapeutics, particularly in the development of vaccines and other immunogenic therapies.14–16 Often, the delivery vehicle itself may play a role in enhancing an immunogenic response,17 stimulating inflammatory processes, and acting as a damage-associated molecular pattern to instigate immune responses.3 Research has demonstrated that the immunogenicity of therapeutics can be significantly influenced by biomaterial delivery vehicle characteristics, such as size, shape, and rigidity,18 which can modulate their interactions with innate immune cells and antigen presenting cells (APC).19–22 As such, the selection of a biomaterial carrier for tolerogenic therapies must be carefully considered to prevent the instigation of an immunogenic immune response. For example, immunotherapies designed to combat autoimmune disease often seek to induce tolerance against one or more autoantigens.23 Use of a biomaterial delivery system capable of activating innate immune cells and APCs toward an inflammatory phenotype runs the risk of instructing the immune system to attack rather than tolerate the antigen, potentially exacerbating disease,24 as may have occurred in an antigen-specific clinical trial for multiple sclerosis.25
As one prominent biomaterial example, poly(lactic-co-glycolic acid) (PLGA) is a widely-used biocompatible and biodegradable polymer approved by the FDA for a variety of uses in parenteral drug delivery,26 and it is ubiquitously used in a micro- or nanoparticulate form to induce either tolerogenic or immunogenic responses. Though PLGA has demonstrated heightened inflammation and APC activation in vivo in the context of large scaffolds27 and microparticles28 in some studies, other microparticle forms have demonstrated the ability to reduce the activation of APCs.29 Indeed, there is some evidence that inherent properties of biomaterials themselves can induce tolerance, even in the absence of antigen specificity or tolerogenic cargo. Several studies have explored the ability of negatively charged microparticles – of numerous materials including PLGA, polystyrene and microdiamond – to bind to positively charged domains of a specific macrophage receptor (MARCO), resulting in sequestration of disease-mediating macrophages away from inflamed tissues in models of multiple sclerosis, encephalitis, and inflammatory bowel disease.20,21,30–32 This illustrates that careful consideration of biomaterial selection in tolerogenic therapy development is critical, complex, and requires further investigation as the field matures.
This review focuses on biomaterials-based approaches to inducing immune tolerance, with a focus on strategies that (1) non-specifically deliver tolerogenic biologics and drugs, (2) deliver antigen with or without biologics or drugs for antigen-specificity, and (3) biomaterial-based modification or delivery of cells to induce tolerance. The benefits and limitations of the three approaches are broadly discussed, as well as their potential for clinical implementation.
2. Biomaterial-based approaches to engineering tolerogenic immunotherapies
Biomaterials can be used to engineer tolerogenic immunotherapies through three broad approaches discussed in this review: tolerogenic biologic or drug delivery, antigen-specific immunotherapy strategies, and tolerogenic cell-based therapies (Fig. 1). These approaches can be applied toward diverse tolerance-related pathologies and disease applications, as summarized in Table 1.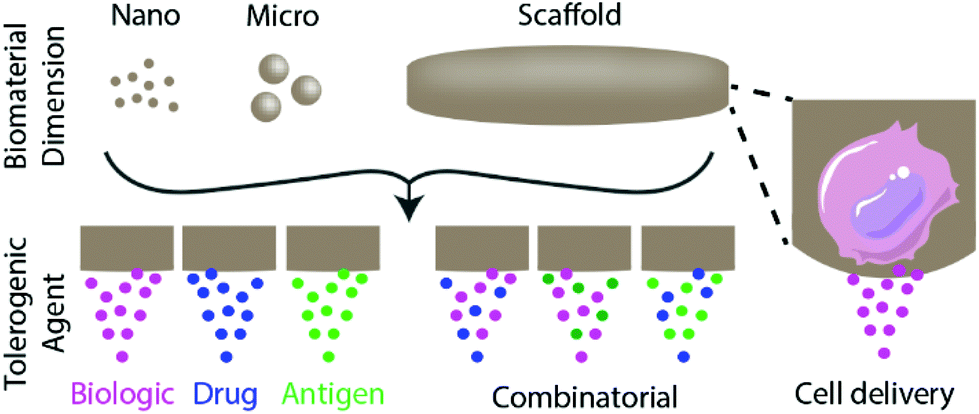 | ||
| Fig. 1 Overview of biomaterial-based approaches to induce immune tolerance discussed in this review. The dimensions of biomaterials used to induce tolerance span nanometre to millimetre length scales. Approaches primarily entail the delivery of tolerogenic biologics and/or drugs, which may include antigens to provide context cues to the immune system or cells to deliver sustained signals. | ||
| Disease and application | Biomaterial | Delivered signals | Ref. |
|---|---|---|---|
| General suppression of immunity | PLGA microparticles | Biologic | 48 |
| PLGA nanoparticles | Biologic | 44 | |
| Drug | 78 | ||
| Modified PLGA nanoparticles | Drug | 87 | |
| Chronic inflammation | PLGA microparticles | Biologic | 49, 50 and 52 |
| Antigen + biologic | 134 | ||
| PLGA nanoparticles | Antigen | 121 and 123 | |
| Multiple sclerosis | PLGA nanoparticles | Drug | 93 |
| Antigen | 110–114 | ||
| Antigen + biologic | 129 | ||
| PLGA microparticles | Antigen | 109 | |
| Antigen + biologic | 133 | ||
| Modified PLGA particles | Antigen | 115 | |
| Antigen + drug | 148, 149 and 155 | ||
| Quantum dots | Antigen | 116 and 117 | |
| PEGylated liposome | Drug | 73 | |
| Gold nanoparticles | Antigen + biologic | 136 | |
| Polyplexes | Antigen + biologic | 139 | |
| Hyaluronic acid | Antigen + biologic | 141–147 | |
| Acetalated dextran microparticles | Antigen + drug | 151 and 152 | |
| Type 1 diabetes | PEG microgels | Biologic | 40 and 41 |
| PLGA microparticles | Antigen + biologic | 130, 131 and 135 | |
| Liposomes | Antigen | 119 | |
| Acetalated dextran microparticles | Antigen + drug | 153 | |
| Chitosan nanoparticles | Antigen + biologic | 137 | |
| Transplant | PLGA nanoparticles | Drug | 81 |
| Antigen | 126–128 | ||
| PLGA scaffold | Biologic | 32 and 35 | |
| PLGA microparticles | Biologic | 51 | |
| PLGA micelles | Drug | 77 and 85 | |
| PEG–PLGA nanoparticles | Drug | 87 | |
| PLA nanoparticles | Drug | 88 | |
| PEG microgels | Drug | 40 and 41 | |
| Modified PEG | Biologic | 64, 67 and 202 | |
| Alginate microgels | Biologic | 65 | |
| PLA/PEG microparticles | Drug + biologic | 101 | |
| Lupus | PLGA nanoparticles | Biologic | 45 |
| Drug | 78 and 80 | ||
| Allergy | PLGA nanoparticles | Antigen | 122 |
| Antigen + biologic | 138 | ||
| Haemophilia A | PLGA nanoparticles | Antigen + drug | 150 |
2.1. Biomaterial-based delivery of tolerogenic biologics or drugs
Biomaterials can be used to deliver and localize tolerogenic biologics or drugs, either to a site of interest or via systemic delivery, to influence a generalized tolerogenic immune response.33 Agents delivered in this manner include proteins (e.g. cytokines and chemokines), RNA, and drugs. In this section, we discuss methods to deliver biologics and drugs via biomaterial systems for a wide range of tolerance-related pathologies.TGF-β1 is a cytokine that plays a pivotal role in maintaining tolerance by inhibiting T cell activation and proliferation and it has demonstrated a role in inducing FOXP3+ Tregs.35 As such, researchers have used localized delivery of this protein to induce tolerance in allogeneic cell transplantation. The Shea group developed TGF-β1-releasing PLGA scaffolds for localized delivery within an islet transplant site for the treatment of type 1 diabetes (T1D).36 They observed a decrease in the expression of inflammatory cytokines from the scaffolds releasing TGF-β1 compared to the control scaffolds, and the transplantation of islets into diabetic mice with TGF-β1-loaded scaffolds resulted in improved blood glucose levels and delayed rejection of allogeneic islets. The Cohen group also explored TGF-β1-presenting scaffolds in the context of allograft rejection, using allofibroblast-seeded microporous alginate scaffolds.37 They observed that cell-seeded scaffolds transplanted in the kidney capsule possessed a higher frequency of immature dendritic cells and Tregs, resulting in an immunoregulatory microenvironment. These studies demonstrate that biomaterial-delivered TGF-β1 has the potential to skew the immune response toward tolerance.
The Shea group also explored localized IL-10 delivery to mediate leukocyte infiltration into porous PLGA scaffolds.38 They used the interesting approach of loading the scaffold with lentivirus to induce sustained IL-10 production in cells infiltrating the scaffold. They found that local IL-10 expression reduced leukocyte infiltration by 50% and reduced localized inflammatory cytokine expression.
The Shea group has also locally delivered the cytokine IL-33 from PLGA scaffolds to induce tolerance in an islet transplant model.39 IL-33 is an immunomodulatory protein that mediates anti-inflammatory properties in adipose tissue and has potentially beneficial effects in cell transplant models.40,41 The Shea group observed that localized delivery of IL-33 from PLGA scaffolds increased the number of enriched Tregs in adipose tissue and extended islet allograft survival but also delayed cell engraftment and function.
Fas ligand (FasL) is a transmembrane protein that induces apoptosis upon binding to the Fas receptor and is a primary mechanism to induce lymphocyte apoptosis.42 Additionally, FasL has been noted to contribute to tumor immune evasion,43 which spurred interest in harnessing this pathway to induce tolerance toward transplanted cells and tissue. The Shirwan and Yolcu group developed a chimeric streptavidin-FasL (SA-FasL) protein that enables the presentation of FasL on biotinylated cells (discussed in depth in section 2.3.1) or biomaterials. In collaboration with the Garcia group, they tethered FasL to the surface of poly(ethylene glycol) (PEG)-based hydrogel microspheres, termed microgels, delivered to the site of transplantation in an allogeneic islet transplant model.44 They observed that PEG microgel-tethered FasL was retained longer at the graft site than soluble FasL and improved allogeneic islet graft survival in diabetic mice, particularly in combination with a short-course, low-dose cover of rapamycin immunosuppression. This group recently employed this strategy with programmed death ligand 1 (PD-L1),45 which has been implicated in tumor immune evasion and shown to reduce T cell proliferation and activation upon ligating its T cell receptor, PD-1.46
Other groups have used combinations of tolerogenic signalling cytokines, such as the co-delivery of TGF-β and IL-2, which are known to synergize to induce Tregs from naïve CD4+ T cells.47 The Fahmy group engineered PLGA nanoparticles loaded with TGF-β and IL-2 that targeted and expanded Tregs.48 They found that this combination of cytokines enhanced Treg stability and reduced pathogenic responses and clinical signs of renal disease in a mouse model of the autoimmune disorder lupus.49
Chemokines are a family of small chemotactic cytokines that control the movement of immune cells during surveillance,50 which makes them a powerful potential tool to manipulate the immune system. CCL22, a macrophage-derived chemokine, has garnered interest for use in tolerogenic therapies due to its ubiquity in tumours and its ability to recruit Tregs that contribute to tumour-specific immune evasion.51 The Little group used localized delivery of CCL22 via degradable PLGA microspheres for a wide range of applications including generalized inflammation,52 periodontal disease and bone loss,53,54 allogeneic transplantation,55 and dry eye disease.56 They demonstrated that sustained CCL22 delivery via PLGA microparticles delivered subcutaneously delays rejection in a hind limb transplant vascular composite allograft model.55 Similarly, they implemented this system to treat periodontal disease, a chronic inflammatory disorder with an autoimmune component that is characterized by bone resorption of the structures supporting the teeth.57 They demonstrated the recruitment of Tregs, which resulted in a complete reversal of the bone loss phenotype.53,54 Another interesting application implemented by this group is in dry eye disease, where the infiltration of CD4+ lymphocytes into the tear film leads to destructive inflammation.58 Local delivery of CCL22-releasing microspheres recruited Tregs and mitigated dry eye disease symptoms by improving tear clearance, corneal epithelial integrity, and goblet cell density.56 Altogether, these studies demonstrate that CCL22 gradients delivered in vivo via degradable microparticles can modulate local immune responses and induce immune tolerance in a wide range of tolerance-related pathologies.
Islet encapsulation using natural or synthetic barriers has long been proposed as a method of reducing or eliminating the need for chronic systemic immunosuppression in allogeneic islet transplantation;59–62 however, encapsulation polymer designs that enable rapid diffusion of insulin (∼7 kDa) inevitably allow the escape of shed donor antigens, which activate the immune response via the indirect antigen recognition pathway.63,64 As such, synergistic tolerogenic approaches are needed to fully eliminate the immune response to encapsulated allogeneic islet grafts. In one such approach, proteins, such as Jagged-1, may be tethered onto the surface of these barriers to provide localized immunomodulation. Jagged-1 is a Notch ligand that interacts with this signalling pathway to alter cell fate decisions.65 Immunosuppression functions are regulated by the overexpression of Jagged-1 through induction, expansion, and differentiation of Tregs and tolerogenic dendritic cells, which is known to induce peripheral tolerance.66,67 In this study, surface immobilization of Jagged-1 on PEGylated islets increased Tregs and regulating cytokine levels in vitro but was not tested long-term in vivo.68
In another approach aiming to synergize with islet encapsulation, the Poznansky group incorporated CXCL12 within alginate capsules for continuous, local release.69 The chemokine CXCL12 can repel effector T cells (Teff) and simultaneously recruit immunosuppressive Tregs.70 They observed that localized delivery of CXCL12 synergized with encapsulation to extend allogeneic and xenogeneic graft survival and increased localized Tregs within the graft.
In a novel approach, the Wang group aimed to induce transplant tolerance by disrupting dendritic cell costimulatory molecule presentation using the CRISPR/Cas9 gene-editing system.71 They used PEG-block-PLGA cationic lipid-assisted nanoparticles to deliver Cas9 mRNA and guide RNA to block the costimulatory molecule CD40 in dendritic cells. They observed that intravenous nanoparticle delivery blocked CD40 in dendritic cells in an acute mouse skin transplant model, reducing graft damage and prolonging graft survival.
Glucocorticoids, such as methylprednisolone, are a widely-used class of anti-inflammatory steroids known to upregulate the secretion of tolerogenic proteins, such as IL-10.76 The Reijerkerk group developed a PEGylated liposome conjugated to the brain-targeting ligand glutathione to enhance the delivery of methylprednisolone to the central nervous system (CNS) for the treatment of multiple sclerosis.77 Multiple sclerosis is a systemic autoimmune disorder characterized by demyelination of the CNS due to the activity of myelin-specific autoreactive T cells.78 They observed that the amount of methylprednisolone taken up by the brain and plasma was increased due to the glutathione PEGylated liposomes, and they demonstrated that treatment with their engineered liposomes was more effective compared to PEG liposomes in experimental autoimmune encephalitis (EAE), a common rodent model of multiple sclerosis.
Another glucocorticoid, betamethasone phosphate, was explored in a biomaterial-based treatment of multiple sclerosis. The Reichardt group developed inorganic–organic hybrid nanoparticles designed for intracellular delivery of glucocorticoid in phagocytic cells.79 They observed that the betamethasone phosphate-loaded nanoparticles primarily modulated macrophages when delivered in vivo and reduced clinical scores in a mouse model of EAE. The Reichardt group also examined the use of their nanoparticle system to treat acute graft-versus-host disease and observed modest improvement in clinical scores over systemic treatment.80
The Pepper group explored the localized delivery of potent glucocorticoid dexamethasone via a micelle delivery vehicle in an allogeneic islet transplant model.81 While micelles alone had a marginal effect on graft survival, this treatment paired with systemic treatment of CTLA-4-Ig, a soluble CD28 antagonist, synergized for 80% graft survival out to 60 days.
Another approach to deliver immunosuppressive drugs via biomaterials is to target critical immune cell subsets, such as professional APCs like dendritic cells. The Fahmy group compared dendritic cell uptake of mycophenolic acid using two nanoparticle delivery systems: a biodegradable PLGA system and a vesicular nanogel platform with a lipid exterior.82 Mycophenolic acid is an immunosuppressive drug used in the treatment of autoimmune disorders and organ transplantation to prevent rejection.83 The nanogel platform was taken up by the dendritic cells more effectively than the PLGA system, and nanogels loaded with mycophenolic acid yielded a greater reduction in inflammatory cytokine production and stimulatory surface marker upregulation than PLGA nanoparticles loaded with mycophenolic acid. Additionally, the nanogel system extended the survival of lupus-prone mice and demonstrated the influence of biomaterial composition on drug uptake by dendritic cells. The Fahmy group further investigated their mycophenolic acid-loaded nanogel delivery system to treat lupus-prone NZB/W F1 mice, and they observed an increase in mouse median survival time by 3 months.84 Dendritic cells that took up the nanogels reduced production of inflammatory cytokines like IFN-γ and IL-12, helping to induce immune tolerance. The Fahmy group also partnered with the Goldstein group to investigate PLGA nanoparticle delivery of mycophenolic acid in an allogeneic mouse skin transplantation model.85 They observed that treatment with drug-loaded nanoparticles resulted in an extension of allograft survival and dendritic cells upregulated PD-L1, resulting in decreased alloreactive T cells.
Rapamycin, commercially known as sirolimus, is a potent immunosuppressant that inhibits T and B lymphocyte activation via mTOR inhibition.86 While potent, systemic delivery of rapamycin results in broad immunosuppression and undesirable side effects; therefore, several groups have explored the use of biomaterials to target its delivery in vivo. The Little group explored a PLGA-based rapamycin delivery system for intracellular delivery in dendritic cells.87 They observed that rapamycin-loaded nanoparticles reduced the ability of the dendritic cells to activate T cells compared to soluble rapamycin. The Samuel group also evaluated the delivery of rapamycin via PLGA nanoparticles on the maturation of dendritic cells.88 They observed that PLGA-encapsulated rapamycin decreased the expression of CD40 and CD86 while increasing the secretion of TGF-β1. In another formulation, the Schnider group engineered an in situ-forming PLGA implant loaded with rapamycin to prevent vascularized composite allotransplantation rejection.89 They observed that treatment with rapamycin-loaded implants prolonged transplant survival and increased the frequency of circulating Tregs.
Cyclosporine A (CsA) is a highly lipophilic peptide used to treat autoimmune diseases and prevent organ transplant rejection by inhibiting T cell activation and proliferation.90 The Cheng group encapsulated CsA in both PEG–PLGA91 and poly(lactic acid) (PLA)92 nanoparticles for targeted suppression in allogeneic transplantation. They observed that free and encapsulated CsA in both PEG–PLGA and PLA nanoparticles reduced T-cell proliferation and inflammatory cytokine production in a dose-dependent manner.
Immunometabolism is an emergent area of immunotherapy, centered around the concept of tuning immune cell function by manipulating cell metabolism.93,94 Much work has pursued this concept in immunogenic applications, but few have exploited this concept in biomaterial-based tolerogenic applications. Histone deacetylase inhibitors (HDACi) are allosteric modulators of metabotropic glutamate receptor 4 (mGluR4), known to regulate transcription of immunomodulatory genes and modulate Treg functions.95 To eliminate side effects associated with long-term systemic use of these drugs, the Little group developed PLGA-based microspheres to deliver HDACi suberoylanilide hydroxamic acid (SAHA) to the lacrimal gland for the treatment of dry eye disease.96 They observed that localized SAHA delivery prevented clinical signs of dry eye disease in mice and reduced the amount of pro-inflammatory cytokines within the lacrimal gland. Additionally, the Jewell group has utilized N-phenyl-7-(hydroxyimino) cyclopropa[b]chromen-1a-carboxamide (PHCCC), a positive allosteric modulator of mGluR4, to manipulate dendritic cell metabolism and skew cytokine secretion toward a regulatory phenotype.97 PHCCC-encapsulated PLGA nanoparticles drastically reduced the toxicity of this drug to dendritic cells in vitro, reduced antigen presentation and activation, and modestly delayed symptom onset in an EAE mouse model, relative to its soluble form, when delivered every 3 days. In a follow-up study, they also evaluated PHCCC delivery using a PEG-modified liposome delivery system.98 They found comparable effects on immune cells in vitro but did not evaluate in vivo efficacy.
Another unique approach to immunomodulation in autoimmune disorders is the targeting of DNA methylation. Generalized hypomethylation of cellular DNA has been linked to autoimmune disorders, though both hypo- and hypermethylated T cells have been identified in lupus patients.99 While systemic treatment with epigenetic modulators could lead to unpredictable outcomes and side effects, there is potential in using biomaterial-based delivery to target specific T cell subsets. The Tsokos and Fahmy groups used 5-azacytidine, a DNA methyltransferase inhibitor delivered via a nanolipogel delivery system tagged with non-depleting CD4- or CD8-specific antibodies in a mouse model of lupus.100 They observed that the delivery of 5-azacytidine to CD4+ T cells promotes FOXP3+ Tregs while CD8+ T cells demonstrated increased cytotoxicity, resulting in an overall reduction in lupus pathology. This team also used their targeted system to deliver calcium/calmodium-dependent protein kinase IV inhibitor KN93, which acts to prevent Th17 cell differentiation.101 They observed that prophylactic targeted delivery of KN93 to CD4+ T cells was more efficient at reducing EAE clinical symptoms than untargeted delivery and reduced lupus pathogenesis at one-tenth of the dose of KN93 systemic delivery.
Lymph node targeting has been employed extensively in immunogenic therapeutic approaches by modulating particle size to target lymphatic drainage.102 The Hubbell group developed PEG-block-poly(propylene sulfide) (PEG-bl-PPS) block copolymer micelles on the order of 50 nm to target lymphatic drainage103 and demonstrated that co-delivery of tacrolimus and rapamycin for 14 consecutive days prolonged allogeneic tail-skin grafts by two-fold.
Anti-drug antibodies have been identified as a contributor to treatment failure in biologic therapy, whereby antibodies generated against biologics inactivate their therapeutic effects.106 To circumvent this reaction, the Maldonado group developed rapamycin-loaded PLGA nanoparticles to induce tolerogenic dendritic cells and prevent hypersensitivity reactions to immunotherapy treatments.107 They observed that the administration of the tolerogenic nanoparticles along with PEGylated hepatic enzyme uricase inhibited the formation of anti-drug antibodies in uricase-deficient mice and non-human primates. They also demonstrated that the tolerogenic nanoparticles loaded with the immunosuppressive drug Adalimumab prevented the formation of anti-drug antibodies against TNFα and prevented arthritis in TNFα transgenic mice.
Other approaches combining biologic and immunosuppressive drugs aim to promote immune tolerance by stably inducing Tregs from naïve T cell populations. The Little group engineered PLGA microparticles for the controlled release of TGF-β1, IL-2, and rapamycin and found they were capable of inducing Tregs in vitro.108 The Little group also investigated the effects of retinoic acid and rapamycin on the number and stability of induced human Tregs.109 They observed that Tregs in the presence of rapamycin demonstrated a potent immunosuppressive effect. The Little group also used this delivery system and biologic/drug combination to mitigate chronic inflammation in dry eye disease, where they found enriched Tregs improved corneal integrity and reduced local inflammatory cytokine concentrations.110
While none of these tolerogenic approaches are in the clinic to our knowledge, biomaterial carriers of drugs or biologics have high translatability, and biomaterial carriers such as PLGA microparticles have been approved for various applications for decades.111 Thus, a clear advantage of non-specific biomaterial-based tolerogenic agent delivery is its relatively high potential for translation in the clinic.
2.2. Antigen-specific biomaterial-based tolerogenic therapy
Biomaterials can also be applied to induce tolerance toward specific antigens in a method akin to vaccination112,113 (Fig. 2). In standard immunogenic vaccines, an antigen specifies the pathogenic target and an adjuvant alerts the immune system to attack. This activates antigen-presenting APCs to generate downstream adaptive immune responses, including B lymphocyte and cytotoxic (CD8+) T lymphocyte activation. By contrast, tolerogenic vaccine therapy involves the delivery of disease- or graft-specific antigens for the treatment of autoimmune disorders and transplantation, respectively. To achieve tolerance rather than immunity against an antigen, two general approaches of antigen presentation by APCs to recipient T cells are used: (1) the delivery or presentation of antigen in the absence of any instructive signalling, with the goal of inducing activation-induced T cell death (AICD) or anergy; and (2) the presentation of antigen with costimulatory signals that instruct T cells toward a tolerogenic or Treg phenotype113 (Fig. 3). Here we discuss approaches to use biomaterials to deliver antigens in both contexts.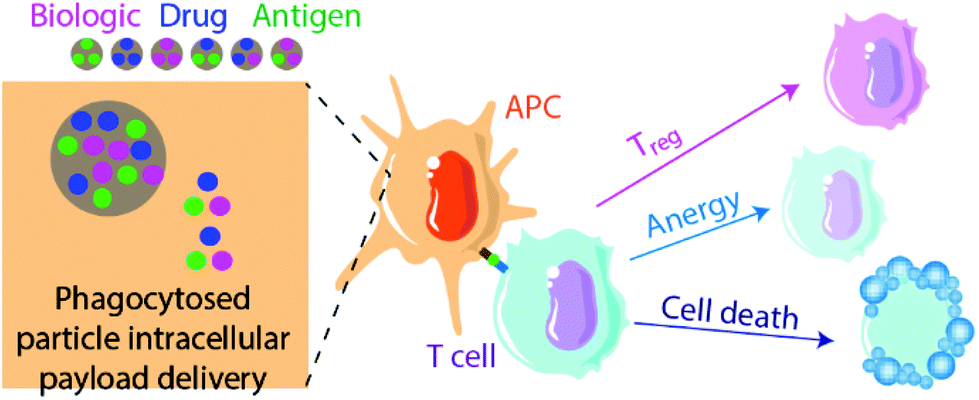 | ||
| Fig. 2 Summary of antigen-specific biomaterial-based approaches to induce tolerance. Typical approaches use nano- or micro-scale particles to deliver either a biologic, drug, antigen payload, or some combination of the three, to influence the interactions of APCs with T cells. Subsequent potential T cell responses include conversion to Tregs, induction of anergy or unresponsiveness, or activation-induced cell death (AICD). | ||
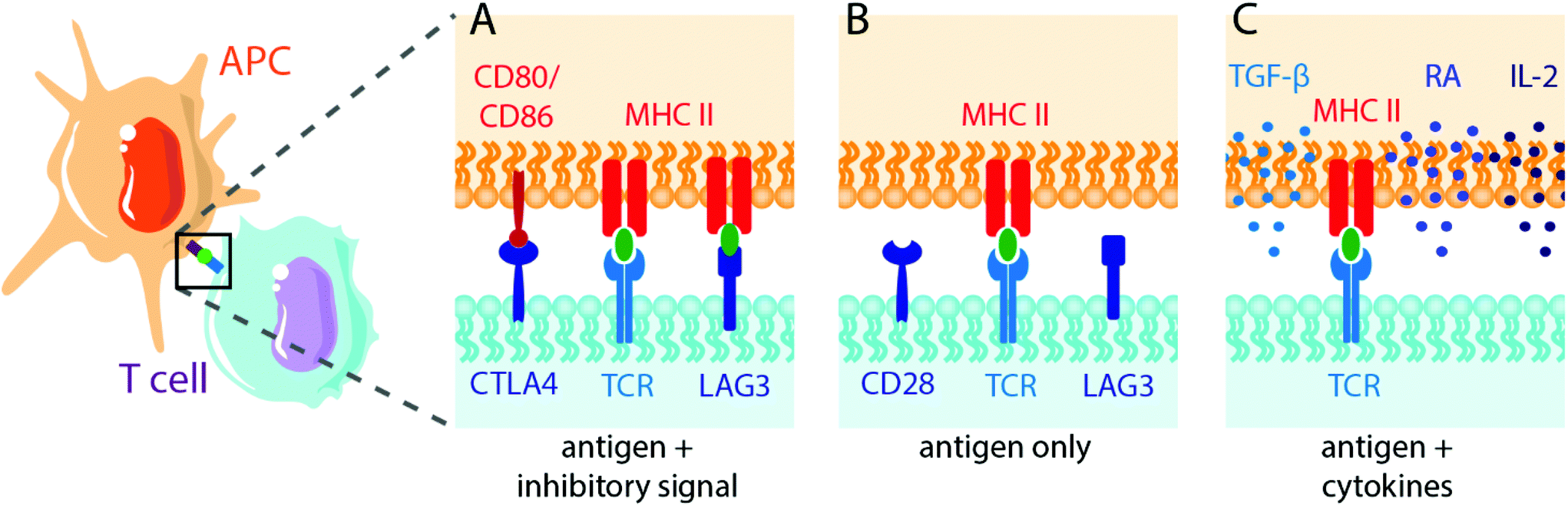 | ||
| Fig. 3 Schematic illustrating antigen-specific APC-T cell interactions to induce T cell tolerance or anergy. APC present antigen to T cell receptors (TCR) via membrane-bound MHC II. Rather than activating co-stimulatory interactions between T cells and APCs, as when pathogens are recognized, tolerance mechanisms involve (A) inhibitory membrane proteins (e.g. LAG3, TIGIT), (B) a lack of secondary signalling (e.g. CD80/CD86), or (C) the presentation of antigen in the presence of tolerogenic cytokines (e.g. IL-2, retinoic acid (RA)). | ||
Multiple sclerosis is one of the most common disease targets in antigen-specific immune therapy and can be readily induced in mice using the EAE model. Myelin oligodendrocyte glycoprotein (MOG), myelin binding protein (MBP) and proteolipid protein (PLP) are commonly used antigen peptides to confer antigen-specific immune tolerance in the studies discussed here.
In one clinically convenient approach, PLGA particles can be used to deliver multiple sclerosis specific MOG or PLP peptides intravenously, and the Miller and Shea group have explored this strategy extensively. In an early study, they found that PLP-coupled PLGA microparticles induced long-term tolerance in an EAE model.114 This was mediated by macrophages expressing the scavenger receptor MARCO, Treg expansion, and T cell abortive activation and anergy. Similarly, they delivered PLP-bearing PLGA nanoparticles intravenously and found reduced CNS infiltration of Th1/Th17 cells and inflammatory monocytes and macrophages.115 They later found that intravenous administration of PLP-encapsulated PLGA nanoparticles induced organ-specific tolerance, as nanoparticles delivered intravenously trafficked to the liver, associated with macrophages, and recruited Ag-specific T cells.116 The tolerance mechanism was independent of the spleen, PD-L1 expression was increased on APCs, and blocking this pathway with PD-1 lessened induced tolerance. They further investigated their PLGA nanoparticle tolerance mechanism when coupled with PLP.117 Ag-Specific T cells cultured in vitro with APCs loaded with PLP-coupled nanoparticles caused reduced T cell proliferation, higher T cell apoptosis, and stronger anti-inflammatory responses. To overcome issues with uncontrollable antigen loading and release, they also investigated the efficacy of direct conjugation of PLP peptides to modified PLGA.118 These nanoparticles had negligible burst release and minimally exposed surface antigen. They found that FOXP3+ Treg induction was dependent on particle size in vitro; however, CD25 expression was not. These PLP-conjugated nanoparticles were effective at inhibiting EAE induced by single or multiple myelin peptides. More recently, this group investigated the effect of particle composition on the efficiency of tolerance induction due to immune cell polarization by comparing encapsulated PLP in low and high molecular weight (MW) PLGA nanoparticles to PLA nanoparticles.119 At low particle doses, PLP-encapsulated PLA particles showed significantly lower clinical scores in an EAE model compared to PLGA-based nanoparticles, with high MW particles showing better clinical scores than low MW particles. PLA particles were associated with Kupffer and liver sinusoidal endothelial cells, which had reduced costimulatory molecule expression correlated with a reduction of CD4+ T cells in the CNS. Interestingly, PLA particles with higher doses of PLP completely ameliorated EAE over 200 days, marked by the inhibition of Th1/Th17 polarization.
In a subcutaneous vaccine-like approach, the Siahaan group developed a PLGA–chitosan–alginate complex to form a colloidal gel, which was used to deliver a modified PLP peptide.120 This modified peptide was engineered to bind to cell surface receptors intercellular adhesion molecule-1 (ICAM-1) and major histocompatibility complex II (MHC II), and the controlled release resulted in EAE suppression and delayed onset, with mechanistic analysis indicating an immune shift away from the Th17 phenotype.
Intravenous approaches to treat or prevent multiple sclerosis have also used non-PLGA nanoparticle formulations with success. The Herkel group used superparamagnetic iron oxide or CdSe/CdS/ZnS-core–shell quantum dot nanoparticles coated in a poly(maleic anhydride-alt-1-octadecane) polymer to intravenously deliver MBP autoantigen.121 They found that their coated particles accumulated selectively in liver sinusoidal epithelial cells, a cell population believed to play a role in hepatic tolerance and to induce the generation of CD4+ FOXP3+ Tregs. This method reversed established EAE and increased splenic Treg populations. The Jewell group has also used MOG-presenting quantum dots and found a ten-fold reduction in the incidence of EAE while investigating the mechanistic influence of MOG surface density on tolerance induction.122
T1D is characterized by the autoimmune destruction of insulin-producing β-cells in the islets of Langerhans, and the non-obese diabetic (NOD) mouse is the most widely used murine model for exploring tolerogenic therapies. Several known autoantigens are associated with the pathology of T1D in both human and mouse versions of the disease, including insulin B peptide, glutamic acid decarboxylase (GAD65), and chromogranin A protein, which is recognized by a population of BDC2.5 CD4+ T cells.123 In one antigen-specific strategy, the Vives-Pi group delivered insulin peptides through phosphatidylserine-liposomes which generated tolerogenic dendritic cells and reduced the autoreactive T cell population to arrest T1D autoimmunity.124 Other groups have used polymer chemistry to alter autoantigens themselves prior to delivery. The Hubbell group has used autoantigen glycosylation to target hepatic APCs, resulting in the prevention of T1D in a NOD model via T cell deletion and anergy, as well as expanded functional Treg populations.125
In another method targeting T1D autoimmunity, the Sanatamaria group used iron oxide nanoparticles coated with peptide-bound MHC complexes.126–128 Monospecific MHC–peptide complexes expanded cognate autoregulatory T cells, prevented disease in pre-diabetic mice and restored normoglycemia in a NOD mouse model. Additionally, using two human autoantigenic epitopes restored normoglycemia in a humanized diabetic mouse model.126 They also showed that this system was capable of expanding antigen-specific regulatory cells in vivo, which were capable of supressing autoantigen-presenting cells and differentiating B cells into regulatory B cells.127 This approach was also effective against primary biliary cholangitis and autoimmune hepatitis in an organ-specific manner and did not supress systemic or local immunity.128
Biomaterial-based delivery of disease-specific antigens has also been applied to other disorders, including celiac disease,129 airway inflammation,130 and rheumatoid arthritis.131 The Getts group intravenously delivered PLGA nanoparticles encapsulating the gliadin protein TIMP-GLIA, derived from wheat, to induce tolerance in mouse models of celiac disease.129 They found decreased immune responsiveness to gliadin, reduced markers of inflammation, and increased gene expression signatures associated with tolerance induction. The Shea and Miller groups have similarly used intravenously-delivered antigen-conjugated polystyrene and PLGA nanoparticles to induce tolerance in an allergic airway inflammation model.130 While antigen-conjugated polystyrene nanoparticles induced tolerance prophylactically, this method led to anaphylaxis in pre-sensitized mice. By contrast, PLGA-encapsulated antigen delivery was well-tolerated and induced tolerance both prophylactically and therapeutically. In an interesting strategy, the Kim group sought to treat collagen-induced arthritis via oral delivery of PLGA nanoparticle-encapsulated collagen II.131 They found that a single oral dose of particles reduced the severity of the disease, suppressed arthritis after disease onset, and was mediated through particle uptake in Peyer's patches.
Antigen-specific immune tolerance induction in allogeneic transplantation aims to use allograft donor antigens, primarily MHC antigens, found on the surface of all nucleated cells.132 In some cases, even when donors are matched to recipient MHC, minor histocompatibility mismatches can cause graft rejection, such as in the case of male-to-female donation by HY peptides encoded on the Y chromosome.133 The Shea group explored the induction of tolerance against the HY peptides Dby and Uty delivered intravenously via PLGA nanoparticles in a sex-mismatched bone marrow transplantation model.134 They found a tolerizing effect with Dby peptide delivered one-day post-transplant, whereas the Uty peptide failed to induce tolerance altogether. In another study, the Shea group sought to induce tolerance against donor MHC in an islet transplant model by coupling antigens to intravenously-delivered PLGA nanoparticles.135 Interestingly, they found tolerance induction in only ∼20% recipients, and it was mediated through the indirect antigen presentation pathway. The effects of this strategy were boosted to ∼60% of recipients when supplemented with a short-course low-dose rapamycin regimen. Similarly, the Shea and Luo groups used their PLGA platform in a fully mismatched murine skin transplant model with a moderate tolerizing effect and they found that both surface conjugation and encapsulation of antigen provided comparable effects.136
The Keselowsky group developed a combinatorial PLGA microparticle delivery system for the simultaneous delivery of disease-specific antigens and immunomodulatory agents.138,139 The approach consists of two particle sizes: (1) phagocytosable particles encapsulating vitamin D3 or insulin B peptide, and (2) nonphagocytosable particles encapsulating TGF-β1 or GM-CSF, delivered subcutaneously in prediabetic NOD mice. Phagocytosable particles targeted dendritic cells, whereas larger particles locally delivered immunosuppressive agents and resulted in a significant reduction in diabetes progression in treated mice. In a similar approach, the Keselowsky group delivered insulin via PLGA microparticles within a GM-CSF- and CpG-containing hydrogel, which resulted in comparable prevention of diabetes onset in NOD mice.140 In partnership with the Avram group, the Keselowsky group adapted this system to deliver MOG in an EAE model.141 This dual-particle approach subcutaneously delivered TGF-β1, vitamin D3, and GM-CSF and resulted in complete protection from disease. Finally, the Lewis group has adapted this system in a model of collagen-induced arthritis.142 Using this same subcutaneous PLGA microparticle delivery system with TGF-β1, vitamin D3, GM-CSF, and disease-specific antigen collagen-II, they demonstrated reduced joint inflammation and expanded Treg populations in joint-proximal lymph nodes after therapeutic administration.
The Mooney group has also used a combination hydrogel-microparticle approach to target autoimmune BDC2.5 CD4+ T cell populations in a NOD T1D model.143 They explored this by either encapsulating BDC peptide (sometimes referred to as P31) in PLGA microparticles embedded in porous alginate GM-CSF-releasing hydrogels, or by tethering BDC peptide to the hydrogel itself. They found that PLGA particle delivery of antigen altered the phenotype of antigen-specific T cells whereas BDC conjugated to alginate polymer generated Tregs in NOD mice, albeit without a significant impact on diabetes progression.
One method to target dendritic cells is via activation of the ligand-activated transcription factor aryl hydrocarbon receptor by 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE). The Quintana group explored this approach using PEG-stabilized gold nanoparticles to co-deliver MOG and ITE in an EAE model.144 They found significant suppression of clinical signs of multiple sclerosis in combinatorial-treated mice but not in mice treated with nanoparticles containing only MOG or ITE.
Another potential method of inducing tolerance in autoimmunity and allergies is via the oral route by targeting DCs in the Peyer's patches of the intestines. The Zong group developed chitosan nanoparticles modified with adhesive ligands, RGD and mannose, loaded with diabetes autoantigen heat shock protein 6.145 The nanoparticles enabled autoantigen stabilization through the digestive tract, targeted to intestinal Peyer's patches, and prevented diabetes in a NOD model out to 23 weeks. In another oral tolerance approach, the Sampson group delivered peanut extract within CpG-coated PLGA nanoparticles in a mouse model of peanut allergy.146 They found decreased levels of allergy-associated cytokines and significant, long-lasting protection from peanut-associated anaphylaxis. This approach is interesting, as it utilizes the well-known potent adjuvant CpG, and seeks to induce Th1 responses to tolerizing the immune system to allergens.
The bacterial DNA sequence CpG is a common adjuvant in immunogenic vaccine design, and the Jewell group developed a unique biomaterial approach with the anti-inflammatory analog, GpG, to generate nanoparticle polyplexes with MOG peptide.147 They found that 3 consecutive doses significantly reduced disease incidence in an EAE model.
The Berkland group has taken an alternative biomaterial approach using hyaluronic acid polymers grafted simultaneously with immune cell-targeting proteins and antigen, which they have termed soluble antigen arrays. In preliminary in vitro studies, they demonstrated that co-grafted ICAM-1, a leukocyte targeting protein, and OVA antigen, reduced T cell activation by DCs in co-culture,148 and modifying this system with PLP in place of OVA successfully suppressed clinical signs of EAE.149,150 In follow-up studies, they grafted hyaluronic acid polymers with PLP and an array of B7-binding peptides to evaluate its efficacy in an EAE model, finding comparable efficacy between B7 receptor and ICAM-1 peptides, with altered cytokine secretion profiles and reduced clinical scores independent of the leukocyte binding protein incorporated into their system.151 In another approach, the Berkland group delivered their soluble antigen arrays conjugated with ICAM-1 and PLP to the lungs for the treatment of EAE.152 Interestingly, PLP delivered alone via this route had substantial efficacy, comparable to soluble antigen arrays, as did bi-functional molecules containing only ICAM-1 tethered to PLP. This antigen-specific formulation has shown comparable efficacy across administration routes, demonstrating its robustness.153 More recent work from this group has investigated the mechanism of interaction of these soluble antigen arrays with B cells in influencing tolerance in EAE models.154,155
The Ainslie group explored the delivery of co-encapsulated drug and antigen within acetalated dextran microparticles.159–161 In one iteration, they co-encapsulated multiple sclerosis-specific MOG peptide with the immunosuppressive glucocorticoid dexamethasone, which has been shown to diminish T cell proliferation and differentiation by attenuating the CD28 co-stimulatory pathway,162 for subcutaneous delivery in an EAE model.159 They found a significant reduction in clinical scores relative to controls after 3 injections. A subsequent study co-encapsulating rapamycin and PLP antigen in acetalated dextran microparticles also showed reduced clinical scores in an EAE model and increased FOXP3 expression, suggesting the expansion of antigen-specific Tregs.160 More recently, the Ainslie group adapted their rapamycin-co-encapsulating microparticle system for the delivery of diabetogenic peptide P31 (also referred to as BDC peptide) in a mouse model of adoptively transferred BDC2.5 CD4+ T cell-induced diabetes.161 Five particle injections post-adoptive transfer, before diabetes symptoms onset, resulted in the prevention of diabetes in the rapamycin/antigen microparticle group only.
The Jewell group also used rapamycin co-delivery with MOG peptide in PLGA microparticles to induce tolerance in an EAE model, using a unique intra-lymph node approach.163 They found that delivery of a single dose at disease peak reduced systemic inflammatory T cells and cytokines and expanded Tregs, resulting in a significant reduction in clinical signs of disease relative to MOG-only and rapamycin-only controls.
Beyond the established risks associated with antigen-specific approaches, antigen-specific immunotherapies face challenges advancing to clinical trials from preclinical models for multiple reasons. First, many preclinical studies demonstrate efficacy in only preventative or prophylactic administration. The onset of autoimmune disorders, such as multiple sclerosis and T1D, are unpredictable and the risk of disease development is challenging to assess, rendering a preventative approach useless, particularly if there is any risk of the approach inducing disease. A few approaches discussed in the section above have demonstrated success in reversing established autoimmune disease, which may make these approaches more feasible to translate. Second, antigen-specific immunotherapies developed in preclinical models are challenging to translate to clinical trials due to variability between autoantigens in preclinical and clinical models of the disease. This is the case for both T1D and multiple sclerosis, where clinical presentation is more complex and less understood than preclinical models, making clinical outcomes difficult to predict.113 Finally, the use of biomaterial delivery systems add complexity to the therapy, introducing safety considerations and potentially requiring independent confirmation of safety of the delivery vehicle via additional study arms. This can increase clinical trial cost, complexity, and patient number, particularly for new and untested biomaterial vehicles. Despite this, at least one clinical trial is in phase I for the antigen-specific treatment of type 1 diabetes, using gold nanoparticle-coupled autoantigens (NCT02837094).
2.3. Tolerogenic cell- and biomaterial-based therapies
An emergent area of biomaterials research is their use to alter or deliver cells as a tolerogenic treatment (Fig. 4). One advantage of cell delivery over biologics or drugs is their potential for more temporally sustained effects, and this was first harnessed for use in immunogenic approaches.165 Biomaterial-based delivery of biologics or drugs is typically temporally limited, where tolerogenic agent delivery is constrained by cargo stability, depot size, and delivery rate.34 By contrast, the delivery of tolerogenic cells may produce more sustained effects, and this can be achieved by targeting tolerant cell subsets, either with ex vivo manipulation or in vivo material targeting.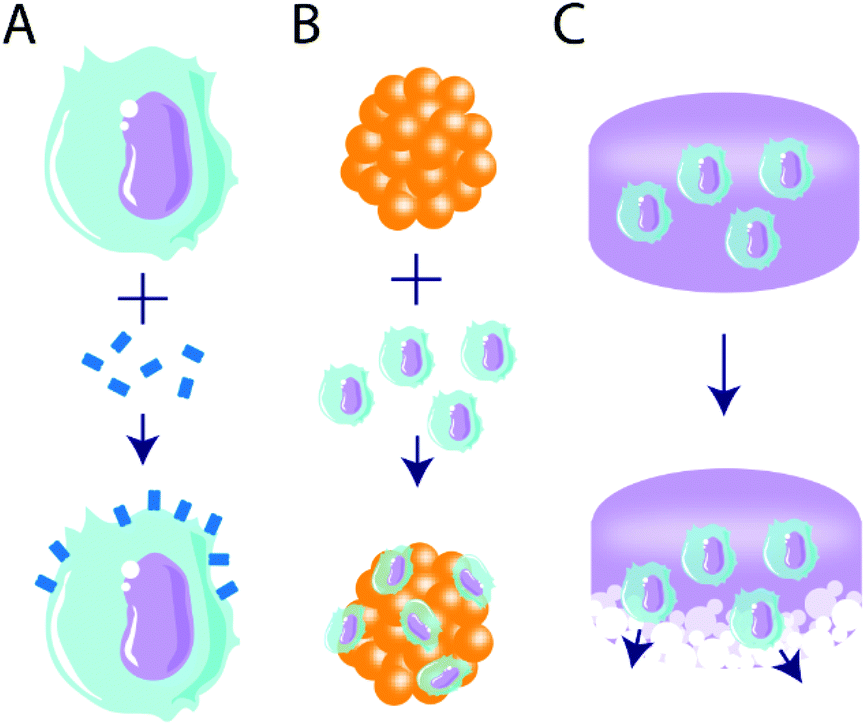 | ||
| Fig. 4 Illustration of tolerogenic combinatorial cell- and biomaterial-based immunotherapeutic strategies. Three primary strategic routes are discussed, including (A) tethering of immunomodulatory molecules to cell surfaces, (B) coating primary cells (e.g. pancreatic islets) with tolerogenic cells, and (C) delivering tolerogenic cells via degradable materials. | ||
Adoptive cell therapy is a method of ex vivo cell manipulation, in which cell subsets of interest are isolated from whole blood, manipulated, expanded, and reintroduced to the patient.166 Though this method has traditionally been used for immune activation in oncology treatments,167,168 tolerant cell types, such as Tregs,169 mesenchymal stem cells (MSC),170–175 and dendritic cells (DC),176 have been targeted to induce immune tolerance. Typical methods include cell isolation, genetic manipulation, and infusion; however, emergent methods make use of biomaterials as a powerful tool to augment or deliver cells to facilitate tolerance induction.
In an islet transplantation model, ECDI-fixed allogeneic splenocytes from BALB/c donor mice were injected into chemically induced diabetic C57BL/6 mice 7 days before and 1 day after islet transplantation.180,181 Islets delivered in PLGA scaffolds alone rejected within 20 days; however, mice that received ECDI-fixed splenocytes showed graft acceptance for the duration of the observation window, 100 days under the kidney capsule180 and 150 days in PLGA scaffolds in the epidydimal fat pad.181 These grafts showed an accumulation of Tregs in the tolerant graft, which prevented CD4+ and CD8+ cells from infiltrating and destroying the islet architecture.181 A multitude of mechanisms was determined to induce this tolerance, including induction of Tregs in lymphoid organs and at the graft site by the donor splenocytes.179
Though multiple sclerosis target-antigens differ between patients and may change over time, commonly targeted antigens such as MBP, MOG, and PLP coupled to splenocytes have been shown to prevent disease and reduce onset and severity after disease induction184–186 by producing antigen-specific tolerance in EAE in vivo models.185,186,188–190 In one study, tolerance was induced to a cocktail of these myelin peptides in an EAE model,190 a strategy that may mediate issues with autoantigen epitope spreading over time. A cocktail of these myelin peptides was coupled to the surface of patient peripheral blood mononuclear cells using EDCI chemistry, then reinfused in the patient within the same day to evaluate procedure safety in a phase 1 clinical trial.182 The results indicated that the therapy was well tolerated, even at higher doses of the myelin peptide-coupled cells where a decrease in T cell-specific responses was seen. More recently, tolerogenic dendritic cells loaded with myelin-derived peptides or aquaporin-4 for multiple sclerosis or neuromyelitis optical spectrum disorders, respectively, were assessed for safety, tolerability, and immunological responses.191 The peptide-loaded dendritic cells were tolerated without adverse side effects and a significant increase of IL-10 levels and Tregs were seen in patients by week 12. These results suggest promising therapies for these peptide-conjugated cells for inflammatory diseases.
In another such approach, erythrocytes were engineered to display disease-relevant antigenic peptides for tolerance induction via a sortase-tagging mechanism.192 Red blood cells (RBC) are consistently cleared from the body through phagocytosis, but they do not show signs of inducing immune responses to their antigens. By exploiting this method of tolerance induction against RBC-displayed antigens, erythrocytes decorated with antigenic peptides can also be presented to induce tolerance. This method demonstrated protection against and reversal of EAE out to 245 days and prevented the onset of T1D in NOD mice out to 210 days.192
We previously discussed the Shirwan group's presentation of FasL on biomaterials to prevent allograft rejection, and they have also used their streptavidin (SA) binding system to modify splenocytes193–196 and pancreatic islets.197,198 SA-FasL-decorated splenocytes inhibited primary and secondary alloreactive responses by blocking proliferative responses and inducing apoptosis of lymphocytes. Long-term persistence of FasL on splenocytes led to the survival of allogeneic islets in a diabetic rat model.196 Pancreatic islets engineered to display SA-FasL on their cell surface normalized blood glucose levels in chemically diabetic syngeneic C57BL/10 mice.197 Viability of the SA-FasL graft was only moderately improved compared to unmodified grafts; however, a short course of rapamycin for 15 days prolonged the modified islet graft indefinitely (>500 days). Specific, localized tolerance was established by inducing local Treg populations in the islet grafts197 through induction of donor antigens and maintenance of Treg cells.198 This approach has also been applied to whole organ transplantations, where SA-FasL tethered to heart endothelial cells allowed for indefinite transplanted heart survival in syngeneic hosts and delayed graft rejection in allogeneic heart grafts.193 Further, allograft survival was extended through the intravenous treatment of graft recipient mice with SA-FasL-decorated splenocytes on days 2 and 6 post-transplantation. Heart grafts that received SA-FasL-decorated splenocytes intraperitoneally on days 1, 3, and 5 post-transplantation with a 15-day daily dose of rapamycin, achieved graft acceptance for the duration of observation (100 days).194,195
The Shirwan and Yolcu team have used the streptavidin–biotin affinity mechanism to decorate cells with other immunomodulatory proteins, such as CD47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 199 and PD-L1.200,201 CD47 protein is an innate immune checkpoint signal that provides “don't-eat-me” self-recognition signals.202 To mitigate islet graft loss via innate immune mechanisms, chimeric SA-CD47 was tethered to islet surfaces without impact on cell viability and function.199 These engineered islets showed better engraftment and function after transplantation via the intraportal hepatic vein, demonstrating low levels of inflammatory cells, including CD11b+ myeloid cells, monocytes, neutrophils, and macrophages, and lower cytokines associated with early islet loss, such as HMGB-1, TF, and IL-1β. Islet surfaces engineered with SA-PD-L1 efficiently converted Teff into Tregs via enhanced TGF-β expression.201 This method had no impact on the viability or function of the islets, and transplantation under the kidney capsule of streptozotocin-induced diabetic C57BL/6 mice demonstrated prolonged survival over islet-only controls out to 40 days. As in previous studies, concurrent short-course rapamycin treatment administered for 15 days post-transplantation resulted in >90% grafts survival and function for the length of the study (100 days). This increase in survival was accompanied by higher levels of regulatory cytokines, such as IDO-1, arginase-1, FOXP3, TGF-β, IL-10, and lower levels of inflammatory cytokines, such as IL-1β, TNF-α, and IFN-γ.200,201 These methods show the potential of streptavidin-engineered proteins and checkpoint inhibitors to promote transplanted cell graft viability and function.
199 and PD-L1.200,201 CD47 protein is an innate immune checkpoint signal that provides “don't-eat-me” self-recognition signals.202 To mitigate islet graft loss via innate immune mechanisms, chimeric SA-CD47 was tethered to islet surfaces without impact on cell viability and function.199 These engineered islets showed better engraftment and function after transplantation via the intraportal hepatic vein, demonstrating low levels of inflammatory cells, including CD11b+ myeloid cells, monocytes, neutrophils, and macrophages, and lower cytokines associated with early islet loss, such as HMGB-1, TF, and IL-1β. Islet surfaces engineered with SA-PD-L1 efficiently converted Teff into Tregs via enhanced TGF-β expression.201 This method had no impact on the viability or function of the islets, and transplantation under the kidney capsule of streptozotocin-induced diabetic C57BL/6 mice demonstrated prolonged survival over islet-only controls out to 40 days. As in previous studies, concurrent short-course rapamycin treatment administered for 15 days post-transplantation resulted in >90% grafts survival and function for the length of the study (100 days). This increase in survival was accompanied by higher levels of regulatory cytokines, such as IDO-1, arginase-1, FOXP3, TGF-β, IL-10, and lower levels of inflammatory cytokines, such as IL-1β, TNF-α, and IFN-γ.200,201 These methods show the potential of streptavidin-engineered proteins and checkpoint inhibitors to promote transplanted cell graft viability and function.
In another innovative method, the Stabler group found that enriched APCs edited to express TGF-β1 on their surfaces using a PEG linker were able to generate functional, antigen-specific Tregs from naïve T cells.176 Though these cells were not tested for their tolerogenic ability in vivo, the Tregs generated were able to suppress the activation of CD4+ T cells in vitro. Additionally, the PEGylation of APCs alone showed suppression of direct antigen-presenting pathways, which may promote immune tolerance. This method could be combined with biomaterial scaffolds to generate Tregs within a transplant site.
Mesenchymal stem or stromal cells (MSC) have also garnered significant interest as a tolerogenic cell type due to their demonstrated potential to suppress activate T cell proliferation.214 Extensive work has explored biomaterials-based delivery of MSCs for regenerative medicine applications,215 with some work targeting these cells for the induction of immune tolerance. Bone marrow-derived MSCs have been evaluated for their immunomodulatory effects on gliadin-specific T cells from celiac patients in vitro.173 MSCs inhibited T cell proliferation and their production of IFN-γ, IL-10, and IL-21, while also reducing CD4+ T cell number and expanding the Treg subset. Baboon MSCs have also demonstrated suppression of allogeneic lymphocyte proliferation in vitro and prolonged skin graft survival moderately through intravenous administration of donor MSCs to MHC-mismatched recipient baboons,174 which may be due to their limited migration.216 MSCs delivered via a PLGA scaffold to a joint in a model of arthritis reduced inflammation and suppressed T cell proliferation.217 Additionally, MSCs delivered in a synthetic, integrin-specific, degradable PEG hydrogel enhanced MSC survival and reparative functions of these cells by modulating Teff functions;175 however, encapsulation of MSCs within biomaterials requires many cells due to low efficiency of engraftment as a large portion of delivered MSCs die or migrate away from the site of transplantation.216
Despite these challenges, cell-based therapies are widely explored in the clinic, with nearly 1200 clinical trials currently underway with MSCs alone, and at least 36 trials delivering MSCs via biomaterial scaffolds, albeit for largely regenerative medicine applications.218 Peptide-coupled cell therapy has been evaluated in a phase 1 clinical trial where it demonstrated good tolerability.182 Unfortunately, cost considerations and manufacturing and regulatory challenges prevented further advancement of this therapy. These challenges are likely to be shared by other cell-based approaches.
3. Future directions for tolerogenic biomaterial-based approaches
The field of tolerogenic biomaterials-based therapies is in its infancy, and several approaches within this review represent new and exciting directions in the field. While only a few researchers have targeted immunometabolism to date, this area has garnered excitement among a few research groups and is likely to expand as it has in the broader field of immunotherapy.94 Similarly, the concept of harnessing cells as tolerogenic tools is in its early stages and is likely to be complemented and enhanced by biomaterial-based approaches to manipulate or deliver cellular therapeutics. Additionally, researchers are investigating immune responses to, and interactions with, biomaterials with ever-increasing complexity,219,220 enabling an approach toward rational biomaterial design and selection for tolerogenic applications that will inevitably further advance the field of tolerogenic immunotherapy. Finally, we must highlight that the majority of the studies discussed within this review have demonstrated great success in the treatment or prevention of tolerogenic pathologies in rodent models. While many immune mechanisms are conserved between rodents and humans, many are not. As such, future studies in this area must approach preclinical immunotherapeutic development with an eye toward clinical translation, perhaps prioritizing in vitro models with human immune cells and tissues or humanized mouse models.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
The authors thank the JDRF for their generous support (1-INO-2020-915-A-N).Notes and references
- P. B. Medawar, Nature, 1961, 189, 14–17 CrossRef CAS.
- R. E. Billingham, L. Brent and P. B. Medawar, Nature, 1953, 172, 603–606 CrossRef CAS.
- S. Sakaguchi, T. Yamaguchi, T. Nomura and M. Ono, Cell, 2008, 133, 775–787 CrossRef CAS.
- K. S. Jones, Semin. Immunol., 2008, 20, 130–136 CrossRef CAS.
- M. Dominguez-Villar and D. A. Hafler, Nat. Immunol., 2018, 19, 665–673 CrossRef CAS.
- M. H. Roberts and E. Erdei, Autoimmun. Rev., 2020, 19, 102423 CrossRef.
- D. Nemazee, Nat. Rev. Immunol., 2017, 17, 281 CrossRef CAS.
- T. Kamradt and N. A. Mitchison, N. Engl. J. Med., 2001, 344, 655–664 CrossRef CAS.
- D. L. Mueller, Nat. Immunol., 2010, 11, 21–27 CrossRef CAS.
- L. Klein, E. A. Robey and C. S. Hsieh, Nat. Rev. Immunol., 2019, 19, 7–18 CrossRef CAS.
- J. M. Gardner, J. J. DeVoss, R. S. Friedman, D. J. Wong, Y. X. Tan, X. Zhou, K. P. Johannes, M. A. Su, H. Y. Chang, M. F. Krummel and M. S. Anderson, Science, 2008, 321, 843–847 CrossRef CAS.
- C. Navarrete and G. F. Bottazzo, Clin. Exp. Immunol., 1993, 91, 189–192 CrossRef CAS.
- K. A. Robinson, W. Orent, J. C. Madsen and G. Benichou, Am. J. Transplant., 2018, 18, 1843–1856 CrossRef.
- M. L. Bookstaver, S. J. Tsai, J. S. Bromberg and C. M. Jewell, Trends Immunol., 2018, 39, 135–150 CrossRef CAS.
- J. M. Gammon, N. M. Dold and C. M. Jewell, Oncotarget, 2016, 7, 15421 CrossRef.
- R. Zhang, M. M. Billingsley and M. J. Mitchell, J. Controlled Release, 2018, 292, 256–276 CrossRef CAS.
- K. S. Jones, Biotechnol. Prog., 2008, 24, 807–814 CrossRef CAS.
- N. Benne, J. van Duijn, J. Kuiper, W. Jiskoot and B. Slütter, J. Controlled Release, 2016, 234, 124–134 CrossRef CAS.
- S. Tomić, V. Kokol, D. Mihajlović, A. Mirčić and M. Čolić, Sci. Rep., 2016, 6, 31618 CrossRef.
- S. Tomić, K. Janjetović, D. Mihajlović, M. Milenković, T. Kravić-Stevović, Z. Marković, B. Todorović-Marković, Z. Spitalsky, M. Micusik, D. Vučević, M. Čolić and V. Trajković, Biomaterials, 2017, 146, 13–28 CrossRef.
- D. R. Getts, R. L. Terry, M. T. Getts, C. Deffrasnes, M. Müller, C. van Vreden, T. M. Ashhurst, B. Chami, D. McCarthy, H. Wu, J. Ma, A. Martin, L. D. Shae, P. Witting, G. S. Kansas, J. Kühn, W. Hafezi, I. L. Campbell, D. Reilly, J. Say, L. Brown, M. Y. White, S. J. Cordwell, S. J. Chadban, E. B. Thorp, S. Bao, S. D. Miller and N. J. C. King, Sci. Transl. Med., 2014, 6, 219ra7 CrossRef.
- R. A. Roberts, T. K. Eitas, J. D. Byrne, B. M. Johnson, P. J. Short, K. P. McKinnon, S. Reisdorf, J. C. Luft, J. M. DeSimone and J. P. Ting, Biomaterials, 2015, 72, 1–10 CrossRef CAS.
- B. T. Kurien and R. H. Scofield, Autoimmun. Rev., 2008, 7, 567–573 CrossRef CAS.
- M. Peakman and C. M. Dayan, Immunology, 2001, 104, 361 CrossRef CAS.
- B. Bielekova, B. Goodwin, N. Richert, I. Cortese, T. Kondo, G. Afshar, B. Gran, J. Eaton, J. Antel and J. A. Frank, Nat. Med., 2000, 6, 1167–1175 CrossRef CAS.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS.
- M. S. Kim, H. H. Ahn, Y. N. Shin, M. H. Cho, G. Khang and H. B. Lee, Biomaterials, 2007, 28, 5137–5143 CrossRef CAS.
- R. Nicolete, D. F. dos Santos and L. H. Faccioli, Int. Immunopharmacol., 2011, 11, 1557–1563 CrossRef CAS.
- R. P. Allen, A. Bolandparvaz, J. A. Ma, V. A. Manickam and J. S. Lewis, ACS Biomater. Sci. Eng., 2018, 4, 900–918 CrossRef CAS.
- S. J. Jeong, J. G. Cooper, I. Ifergan, T. L. McGuire, D. Xu, Z. Hunter, S. Sharma, D. McCarthy, S. D. Miller and J. A. Kessler, Neurobiol. Dis., 2017, 108, 73–82 CrossRef CAS.
- V. Volarevic, V. Paunovic, Z. Markovic, B. Simovic Markovic, M. Misirkic-Marjanovic, B. Todorovic-Markovic, S. Bojic, L. Vucicevic, S. Jovanovic and N. Arsenijevic, ACS Nano, 2014, 8, 12098–12109 CrossRef CAS.
- J. Tosic, Z. Stanojevic, S. Vidicevic, A. Isakovic, D. Ciric, T. Martinovic, T. Kravic-Stevovic, V. Bumbasirevic, V. Paunovic and S. Jovanovic, Neuropharmacology, 2019, 146, 95–108 CrossRef CAS.
- J. I. Andorko, K. L. Hess and C. M. Jewell, AAPS J., 2015, 17, 323–338 CrossRef CAS.
- R. Langer and N. A. Peppas, AIChE J., 2003, 49, 2990–3006 CrossRef CAS.
- D. A. Clark and R. Coker, Int. J. Biochem. Cell Biol., 1998, 30, 293–298 CrossRef CAS.
- J. M. Liu, J. Zhang, X. Zhang, K. A. Hlavaty, C. F. Ricci, J. N. Leonard, L. D. Shea and R. M. Gower, Biomaterials, 2016, 80, 11–19 CrossRef CAS.
- S. Orr, I. Strominger, E. Eremenko, E. Vinogradov, E. Ruvinov, A. Monsonego and S. Cohen, Acta Biomater., 2016, 45, 196–209 CrossRef CAS.
- R. M. Gower, R. M. Boehler, S. M. Azarin, C. F. Ricci, J. N. Leonard and L. D. Shea, Biomaterials, 2014, 35, 2024–2031 CrossRef CAS.
- J. M. Liu, X. Zhang, S. Joe, X. Luo and L. D. Shea, J. Immunol. Regener. Med., 2018, 1, 1–12 CrossRef.
- Q. Liu and H. R. Turnquist, Cytokine, 2013, 62, 183–194 CrossRef CAS.
- B. M. Matta and H. R. Turnquist, Methods Mol. Biol., 2016, 1371, 29–41 CrossRef CAS.
- T. S. Griffith, T. Brunner, S. M. Fletcher, D. R. Green and T. A. Ferguson, Science, 1995, 270, 1189–1192 CrossRef CAS.
- B. C. Barnhart, P. Legembre, E. Pietras, C. Bubici, G. Franzoso and M. E. Peter, EMBO J., 2004, 23, 3175–3185 CrossRef CAS.
- D. M. Headen, K. B. Woodward, M. M. Coronel, P. Shrestha, J. D. Weaver, H. Zhao, M. Tan, M. D. Hunckler, W. S. Bowen, C. T. Johnson, L. Shea, E. S. Yolcu, A. J. García and H. Shirwan, Nat. Mater., 2018, 17, 732–739 CrossRef CAS.
- M. M. Coronel, K. E. Martin, M. D. Hunckler, G. Barber, E. B. O'Neill, J. D. Medina, E. Opri, C. A. McClain, L. Batra and J. D. Weaver, Sci. Adv., 2020, 6, eaba5573 CrossRef CAS.
- C. Blank, T. F. Gajewski and A. Mackensen, Cancer Immunol. Immunother., 2005, 54, 307–314 CrossRef CAS.
- C. I. Kingsley, M. Karim, A. R. Bushell and K. J. Wood, J. Immunol., 2002, 168, 1080–1086 CrossRef CAS.
- M. D. McHugh, J. Park, R. Uhrich, W. Gao, D. A. Horwitz and T. M. Fahmy, Biomaterials, 2015, 59, 172–181 CrossRef CAS.
- D. A. Horwitz, S. Bickerton, M. Koss, T. M. Fahmy and A. L. Cava, Arthritis Rheumatol., 2019, 71, 632–640 CrossRef CAS.
- M. Baggiolini, Nature, 1998, 392, 565–568 CrossRef CAS.
- U. Yamashita and E. Kuroda, Crit. Rev. Immunol., 2002, 22, 105–114 CAS.
- S. Jhunjhunwala, G. Raimondi, A. J. Glowacki, S. J. Hall, D. Maskarinec, S. H. Thorne, A. W. Thomson and S. R. Little, Adv. Mater., 2012, 24, 4735–4738 CrossRef CAS.
- A. C. Araujo-Pires, A. E. Vieira, C. F. Francisconi, C. C. Biguetti, A. Glowacki, S. Yoshizawa, A. P. Campanelli, A. P. F. Trombone, C. S. Sfeir and S. R. Little, J. Bone Miner. Res., 2015, 30, 412–422 CrossRef.
- A. J. Glowacki, S. Yoshizawa, S. Jhunjhunwala, A. E. Vieira, G. P. Garlet, C. Sfeir and S. R. Little, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18525–18530 CrossRef CAS.
- J. D. Fisher, W. Zhang, S. C. Balmert, A. M. Aral, A. P. Acharya, Y. Kulahci, J. Li, H. R. Turnquist, A. W. Thomson and M. G. Solari, Sci. Adv., 2020, 6, eaax8429 CrossRef CAS.
- M. L. Ratay, A. J. Glowacki, S. C. Balmert, A. P. Acharya, J. Polat, L. P. Andrews, M. V. Fedorchak, J. S. Schuman, D. A. Vignali and S. R. Little, J. Controlled Release, 2017, 258, 208–217 CrossRef CAS.
- R. C. Williams, N. Engl. J. Med., 1990, 322, 373–382 CrossRef CAS.
- J. L. Gayton, Clin. Ophthalmol., 2009, 3, 405–412 CrossRef.
- T. Desai and L. D. Shea, Nat. Rev. Drug Discovery, 2017, 16, 338–350 CrossRef CAS.
- J. A. Giraldo, J. D. Weaver and C. L. Stabler, J. Diabetes Sci. Technol., 2010, 4, 1238–1247 CrossRef.
- A. D. Salama, K. L. Womer and M. H. Sayegh, J. Immunol., 2007, 178, 5419–5423 CrossRef CAS.
- J. L. Zakrzewski, M. R. M. van den Brink and J. A. Hubbell, Nat. Biotechnol., 2014, 32, 786–794 CrossRef CAS.
- Y. Li, A. W. Frei, E. Y. Yang, I. Labrada-Miravet, C. Sun, Y. Rong, M. M. Samojlik, A. L. Bayer and C. L. Stabler, Biomaterials, 2020, 256, 120182 CrossRef CAS.
- J. D. Weaver, D. M. Headen, M. M. Coronel, M. D. Hunckler, H. Shirwan and A. J. García, Am. J. Transplant., 2019, 19, 1315–1327 CrossRef CAS.
- A. M. Grochowski, K. M. Loomes and N. B. Spinner, Gene, 2016, 576, 381–384 CrossRef.
- E. F. Cahill, L. M. Tobin, F. Carty, B. P. Mahon and K. English, Stem Cell Res. Ther., 2015, 6, 19 CrossRef.
- E. S. Yvon, S. Vigouroux, R. F. Rousseau, E. Biagi, P. Amrolia, G. Dotti, H. J. Wagner and M. K. Brenner, Blood, 2003, 102, 3815–3821 CrossRef CAS.
- Z. Izadi, E. Hajizadeh-Saffar, J. Hadjati, M. Habibi-Anbouhi, M. H. Ghanian, H. Sadeghi-Abandansari, M. K. Ashtiani, Z. Samsonchi, M. Raoufi, M. Moazenchi, M. Izadi, A. S. S. H. Nejad, H. Namdari, Y. Tahamtani, S. N. Ostad, H. Akbari-Javar and H. Baharvand, Biomaterials, 2018, 182, 191–201 CrossRef CAS.
- T. Chen, J. Yuan, S. Duncanson, M. L. Hibert, B. C. Kodish, G. Mylavaganam, M. Maker, H. Li, M. Sremac, M. Santosuosso, B. Forbes, S. Kashiwagi, J. Cao, J. Lei, M. Thomas, C. Hartono, D. Sachs, J. Markmann, A. Sambanis and M. C. Poznansky, Am. J. Transplant., 2015, 15, 618–627 CrossRef CAS.
- L. Zou, B. Barnett, H. Safah, V. F. LaRussa, M. Evdemon-Hogan, P. Mottram, S. Wei, O. David, T. J. Curiel and W. Zou, Cancer Res., 2004, 64, 8451–8455 CrossRef CAS.
- Y. Zhang, S. Shen, G. Zhao, C. F. Xu, H. B. Zhang, Y. L. Luo, Z. T. Cao, J. Shi, Z. B. Zhao, Z. X. Lian and J. Wang, Biomaterials, 2019, 217, 119302 CrossRef CAS.
- J. Esdaile, L. Joseph, T. MacKenzie, M. Kashgarian and J. Hayslett, J. Rheumatol., 1994, 21, 2046–2051 CAS.
- M. Zaffaroni, A. Ghezzi and G. Comi, Neurol. Sci., 2006, 27, s13–s17 CrossRef.
- K. C. Meyer, C. Decker and R. Baughman, Clin. Chest Med., 2010, 31, 565–588 CrossRef.
- S. J. Rossi, T. J. Schroeder, S. Hariharan and M. Roy First, Drug Saf., 1993, 9, 104–131 CrossRef CAS.
- P. J. Barnes, Clin. Sci., 1998, 94, 557–572 CrossRef CAS.
- P. J. Gaillard, C. C. M. Appeldoorn, J. Rip, R. Dorland, S. M. A. van der Pol, G. Kooij, H. E. de Vries and A. Reijerkerk, J. Controlled Release, 2012, 164, 364–369 CrossRef CAS.
- C. Baecher-Allan, B. J. Kaskow and H. L. Weiner, Neuron, 2018, 97, 742–768 CrossRef CAS.
- E. Montes-Cobos, S. Ring, H. J. Fischer, J. Heck, J. Strauß, M. Schwaninger, S. D. Reichardt, C. Feldmann, F. Lühder and H. M. Reichardt, J. Controlled Release, 2017, 245, 157–169 CrossRef CAS.
- T. K. Kaiser, H. Li, L. Roßmann, S. D. Reichardt, H. Bohnenberger, C. Feldmann and H. M. Reichardt, Eur. J. Immunol., 2020, 50, 1220–1233 CrossRef CAS.
- P. Kuppan, S. Kelly, K. Polishevska, O. Hojanepesov, K. Seeberger, G. S. Korbutt and A. R. Pepper, Am. J. Transplant., 2020, 20, 714–725 CrossRef CAS.
- M. Look, W. M. Saltzman, J. Craft and T. M. Fahmy, Biomaterials, 2014, 35, 1089–1095 CrossRef CAS.
- T. J. Franklin and J. M. Cook, Biochem. J., 1969, 113, 515–524 CrossRef CAS.
- M. Look, E. Stern, Q. A. Wang, L. D. DiPlacido, M. Kashgarian, J. Craft and T. M. Fahmy, J. Clin. Invest., 2013, 123, 1741–1749 CrossRef CAS.
- A. C. Shirali, M. Look, W. Du, E. Kassis, H. W. Stout-Delgado, T. M. Fahmy and D. R. Goldstein, Am. J. Transplant., 2011, 11, 2582–2592 CrossRef CAS.
- J. Li, S. G. Kim and J. Blenis, Cell Metab., 2014, 19, 373–379 CrossRef CAS.
- S. Jhunjhunwala, G. Raimondi, A. W. Thomson and S. R. Little, J. Controlled Release, 2009, 133, 191–197 CrossRef CAS.
- A. Haddadi, P. Elamanchili, A. Lavasanifar, S. Das, J. Shapiro and J. Samuel, J. Biomed. Mater. Res., Part A, 2008, 84, 885–898 CrossRef.
- D. Sutter, D. V. Dzhonova, J. C. Prost, C. Bovet, Y. Banz, L. Rahnfeld, J. C. Leroux, R. Rieben, E. Vögelin, J. A. Plock, P. Luciani, A. Taddeo and J. T. Schnider, Sci. Rep., 2019, 9, 9269 CrossRef.
- A. D. Hess, A. H. Esa and P. M. Colombani, Transplant. Proc., 1988, 20, 29–40 CAS.
- L. Tang, J. Azzi, M. Kwon, M. Mounayar, R. Tong, Q. Yin, R. Moore, N. Skartsis, T. M. Fan and R. Abdi, J. Transplant., 2012, 2012, 896141 Search PubMed.
- J. Azzi, L. Tang, R. Moore, R. Tong, N. E. Haddad, T. Akiyoshi, B. Mfarrej, S. Yang, M. Jurewicz, T. Ichimura, N. Lindeman, J. Cheng and R. Abdi, FASEB J., 2010, 24, 3927–3938 CrossRef CAS.
- R. Newton, B. Priyadharshini and L. A. Turka, Nat. Immunol., 2016, 17, 618 CrossRef CAS.
- L. A. O'Neill, R. J. Kishton and J. Rathmell, Nat. Rev. Immunol., 2016, 16, 553 CrossRef.
- T. Akimova, G. Ge, T. Golovina, T. Mikheeva, L. Wang, J. L. Riley and W. W. Hancock, Clin. Immunol., 2010, 136, 348–363 CrossRef CAS.
- M. L. Ratay, S. C. Balmert, E. J. Bassin and S. R. Little, Acta Biomater., 2018, 71, 261–270 CrossRef CAS.
- J. M. Gammon, L. H. Tostanoski, A. R. Adapa, Y. C. Chiu and C. M. Jewell, J. Controlled Release, 2015, 210, 169–178 CrossRef CAS.
- J. M. Gammon, A. R. Adapa and C. M. Jewell, J. Biomed. Mater. Res., Part A, 2017, 105, 2977–2985 CrossRef CAS.
- N. Altorok and A. H. Sawalha, Curr. Opin. Rheumatol., 2013, 25, 569–576 CrossRef CAS.
- H. Li, M. G. Tsokos, S. Bickerton, A. Sharabi, Y. Li, V. R. Moulton, P. Kong, T. M. Fahmy and G. C. Tsokos, JCI Insight, 2018, 3, e120880 CrossRef.
- K. Otomo, T. Koga, M. Mizui, N. Yoshida, C. Kriegel, S. Bickerton, T. M. Fahmy and G. C. Tsokos, J. Immunol., 2015, 195, 5533–5537 CrossRef CAS.
- F. Ikomi, G. K. Hanna and G. W. Schmid-Schonbein, Lymphology, 1999, 32, 90–102 CAS.
- K. Y. Dane, C. Nembrini, A. A. Tomei, J. K. Eby, C. P. O'Neil, D. Velluto, M. A. Swartz, L. Inverardi and J. A. Hubbell, J. Controlled Release, 2011, 156, 154–160 CrossRef CAS.
- P. R. Streeter, B. T. Rouse and E. C. Butcher, J. Cell Biol., 1988, 107, 1853–1862 CrossRef CAS.
- J. Azzi, Q. Yin, M. Uehara, S. Ohori, L. Tang, K. Cai, T. Ichimura, M. McGrath, O. Maarouf and E. Kefaloyianni, Cell Rep., 2016, 15, 1202–1213 CrossRef CAS.
- V. Strand, A. Balsa, J. Al-Saleh, L. Barile-Fabris, T. Horiuchi, T. Takeuchi, S. Lula, C. Hawes, B. Kola and L. Marshall, BioDrugs, 2017, 31, 299–316 CrossRef CAS.
- T. K. Kishimoto, J. D. Ferrari, R. A. LaMothe, P. N. Kolte, A. P. Griset, C. O'Neil, V. Chan, E. Browning, A. Chalishazar and W. Kuhlman, Nat. Nanotechnol., 2016, 11, 890–899 CrossRef CAS.
- S. Jhunjhunwala, S. C. Balmert, G. Raimondi, E. Dons, E. E. Nichols, A. W. Thomson and S. R. Little, J. Controlled Release, 2012, 159, 78–84 CrossRef CAS.
- S. Jhunjhunwala, L. C. Chen, E. E. Nichols, A. W. Thomson, G. Raimondi and S. R. Little, J. Leukocyte Biol., 2013, 94, 981–989 CrossRef CAS.
- M. L. Ratay, S. C. Balmert, A. P. Acharya, A. C. Greene, T. Meyyappan and S. R. Little, Sci. Rep., 2017, 7, 17527 CrossRef.
- J. S. Choi, K. Seo and J. W. Yoo, J. Pharm. Invest., 2012, 42, 155–163 CrossRef CAS.
- R. M. Pearson, L. M. Casey, K. R. Hughes, S. D. Miller and L. D. Shea, Adv. Drug Delivery Rev., 2017, 114, 240–255 CrossRef CAS.
- S. D. Miller, D. M. Turley and J. R. Podojil, Nat. Rev. Immunol., 2007, 7, 665–677 CrossRef CAS.
- D. R. Getts, A. J. Martin, D. P. McCarthy, R. L. Terry, Z. N. Hunter, W. T. Yap, M. T. Getts, M. Pleiss, X. Luo, N. J. King, L. D. Shea and S. D. Miller, Nat. Biotechnol., 2012, 30, 1217–1224 CrossRef CAS.
- Z. Hunter, D. P. McCarthy, W. T. Yap, C. T. Harp, D. R. Getts, L. D. Shea and S. D. Miller, ACS Nano, 2014, 8, 2148–2160 CrossRef CAS.
- D. P. McCarthy, J. W. T. Yap, C. T. Harp, W. K. Song, J. Chen, R. M. Pearson, S. D. Miller and L. D. Shea, Nanomedicine, 2017, 13, 191–200 CrossRef CAS.
- R. Kuo, E. Saito, S. D. Miller and L. D. Shea, Mol. Ther., 2017, 25, 1676–1685 CrossRef CAS.
- R. M. Pearson, L. M. Casey, K. R. Hughes, L. Z. Wang, M. G. North, D. R. Getts, S. D. Miller and L. D. Shea, Mol. Ther., 2017, 25, 1655–1664 CrossRef CAS.
- E. Saito, R. Kuo, K. R. Kramer, N. Gohel, D. A. Giles, B. B. Moore, S. D. Miller and L. D. Shea, Biomaterials, 2019, 222, 119432 CrossRef CAS.
- B. Büyüktimkin, Q. Wang, P. Kiptoo, J. M. Stewart, C. Berkland and T. J. Siahaan, Mol. Pharm., 2012, 9, 979–985 CrossRef.
- A. Carambia, B. Freund, D. Schwinge, O. T. Bruns, S. C. Salmen, H. Ittrich, R. Reimer, M. Heine, S. Huber, C. Waurisch, A. Eychmüller, D. C. Wraith, T. Korn, P. Nielsen, H. Weller, C. Schramm, S. Lüth, A. W. Lohse, J. Heeren and J. Herkel, J. Hepatol., 2015, 62, 1349–1356 CrossRef CAS.
- K. L. Hess, E. Oh, L. H. Tostanoski, J. I. Andorko, K. Susumu, J. R. Deschamps, I. L. Medintz and C. M. Jewell, Adv. Funct. Mater., 2017, 27, 1700290 CrossRef.
- C. E. Taplin and J. M. Barker, Autoimmunity, 2008, 41, 11–18 CrossRef CAS.
- I. Pujol-Autonell, A. Serracant-Prat, M. Cano-Sarabia, R. M. Ampudia, S. Rodriguez-Fernandez, A. Sanchez, C. Izquierdo, T. Stratmann, M. Puig-Domingo, D. Maspoch, J. Verdaguer and M. Vives-Pi, PLoS One, 2015, 10, e0127057 CrossRef.
- D. S. Wilson, M. Damo, S. Hirosue, M. M. Raczy, K. Brünggel, G. Diaceri, X. Quaglia-Thermes and J. A. Hubbell, Nat. Biomed. Eng., 2019, 3, 817–829 CrossRef CAS.
- S. Tsai, A. Shameli, J. Yamanouchi, X. Clemente-Casares, J. Wang, P. Serra, Y. Yang, Z. Medarova, A. Moore and P. Santamaria, Immunity, 2010, 32, 568–580 CrossRef CAS.
- X. Clemente-Casares, J. Blanco, P. Ambalavanan, J. Yamanouchi, S. Singha, C. Fandos, S. Tsai, J. Wang, N. Garabatos, C. Izquierdo, S. Agrawal, M. B. Keough, V. W. Yong, E. James, A. Moore, Y. Yang, T. Stratmann, P. Serra and P. Santamaria, Nature, 2016, 530, 434–440 CrossRef CAS.
- C. S. Umeshappa, S. Singha, J. Blanco, K. Shao, R. H. Nanjundappa, J. Yamanouchi, A. Parés, P. Serra, Y. Yang and P. Santamaria, Nat. Commun., 2019, 10, 2150 CrossRef.
- T. L. Freitag, J. R. Podojil, R. M. Pearson, F. J. Fokta, C. Sahl, M. Messing, L. C. Andersson, K. Leskinen, P. Saavalainen, L. I. Hoover, K. Huang, D. Phippard, S. Maleki, N. J. C. King, L. D. Shea, S. D. Miller, S. K. Meri and D. R. Getts, Gastroenterology, 2020, 158, 1667–1681 CrossRef CAS.
- C. B. Smarr, W. T. Yap, T. P. Neef, R. M. Pearson, Z. N. Hunter, I. Ifergan, D. R. Getts, P. J. Bryce, L. D. Shea and S. D. Miller, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5059–5064 CrossRef CAS.
- W. U. Kim, W. K. Lee, J. W. Ryoo, S. H. Kim, J. Kim, J. Youn, S. Y. Min, E. Y. Bae, S. Y. Hwang, S. H. Park, C. S. Cho, J. S. Park and H. Y. Kim, Arthritis Rheum., 2002, 46, 1109–1120 CrossRef CAS.
- M. Sykes, J. Intern. Med., 2007, 262, 288–310 CrossRef CAS.
- W. Wang, L. R. Meadows, J. M. M. den Haan, N. E. Sherman, Y. Chen, E. Blokland, J. Shabanowitz, A. I. Agulnik, R. C. Hendrickson, C. E. Bishop, D. F. Hunt, E. Goulmy and V. H. Engelhard, Science, 1995, 269, 1588–1590 CrossRef CAS.
- K. A. Hlavaty, D. P. McCarthy, E. Saito, W. T. Yap, S. D. Miller and L. D. Shea, Biomaterials, 2016, 76, 1–10 CrossRef CAS.
- J. Bryant, K. A. Hlavaty, X. Zhang, W. T. Yap, L. Zhang, L. D. Shea and X. Luo, Biomaterials, 2014, 35, 8887–8894 CrossRef CAS.
- S. Shah, S. Daneshmandi, K. R. Hughes, S. Yu, A. M. Bedoya, L. D. Shea and X. Luo, Biomaterials, 2019, 210, 70–82 CrossRef CAS.
- L. M. Casey, R. M. Pearson, K. R. Hughes, J. M. H. Liu, J. A. Rose, M. G. North, L. Z. Wang, M. Lei, S. D. Miller and L. D. Shea, Bioconjugate Chem., 2018, 29, 813–823 CrossRef CAS.
- J. S. Lewis, N. V. Dolgova, Y. Zhang, C. Q. Xia, C. H. Wasserfall, M. A. Atkinson, M. J. Clare-Salzler and B. G. Keselowsky, Clin. Immunol., 2015, 160, 90–102 CrossRef CAS.
- J. S. Lewis, J. M. Stewart, G. P. Marshall, M. R. Carstens, Y. Zhang, N. V. Dolgova, C. Xia, T. M. Brusko, C. H. Wasserfall, M. J. Clare-Salzler, M. A. Atkinson and B. G. Keselowsky, ACS Biomater. Sci. Eng., 2019, 5, 2631–2646 CrossRef CAS.
- Y. M. Yoon, J. S. Lewis, M. R. Carstens, M. Campbell-Thompson, C. H. Wasserfall, M. A. Atkinson and B. G. Keselowsky, Sci. Rep., 2015, 5, 13155 CrossRef CAS.
- J. J. Cho, J. M. Stewart, T. T. Drashansky, M. A. Brusko, A. N. Zuniga, K. J. Lorentsen, B. G. Keselowsky and D. Avram, Biomaterials, 2017, 143, 79–92 CrossRef CAS.
- R. Allen, S. Chizari, J. A. Ma, S. Raychaudhuri and J. S. Lewis, ACS Appl. Bio Mater., 2019, 2, 2388–2404 CrossRef.
- C. S. Verbeke, S. Gordo, D. A. Schubert, S. A. Lewin, R. M. Desai, J. Dobbins, K. W. Wucherpfennig and D. J. Mooney, Adv. Healthcare Mater., 2017, 6, 1600773 CrossRef.
- A. Yeste, M. Nadeau, E. J. Burns, H. L. Weiner and F. J. Quintana, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11270–11275 CrossRef CAS.
- Y. Chen, J. Wu, J. Wang, W. Zhang, B. Xu, X. Xu and L. Zong, Diabetologia, 2018, 61, 1384–1396 CrossRef CAS.
- K. D. Srivastava, A. Siefert, T. M. Fahmy, M. J. Caplan, X. M. Li and H. A. Sampson, J. Allergy Clin. Immunol., 2016, 138, 536–543 CrossRef CAS.
- K. L. Hess, J. I. Andorko, L. H. Tostanoski and C. M. Jewell, Biomaterials, 2017, 118, 51–62 CrossRef CAS.
- C. Chittasupho, J. Sestak, L. Shannon, T. J. Siahaan, C. M. Vines and C. Berkland, Mol. Pharm., 2014, 11, 367–373 CrossRef CAS.
- J. O. Sestak, B. P. Sullivan, S. Thati, L. Northrup, B. Hartwell, L. Antunez, M. L. Forrest, C. M. Vines, T. J. Siahaan and C. Berkland, Mol. Ther.–Methods Clin. Dev., 2014, 1, 14008 CrossRef.
- J. Sestak, M. Mullins, L. Northrup, S. Thati, M. L. Forrest, T. J. Siahaan and C. Berkland, J. Controlled Release, 2013, 168, 334–340 CrossRef CAS.
- L. Northrup, J. O. Sestak, B. P. Sullivan, S. Thati, B. L. Hartwell, T. J. Siahaan, C. M. Vines and C. Berkland, AAPS J., 2014, 16, 1204–1213 CrossRef CAS.
- C. Kuehl, S. Thati, B. Sullivan, J. Sestak, M. Thompson, T. Siahaan and C. Berkland, J. Pharm. Sci., 2017, 106, 3293–3302 CrossRef CAS.
- S. Thati, C. Kuehl, B. Hartwell, J. Sestak, T. Siahaan, L. Forrest and C. Berkland, J. Pharm. Sci., 2015, 104, 714–721 CrossRef CAS.
- B. L. Hartwell, C. J. Pickens, M. Leon and C. Berkland, Biomacromolecules, 2017, 18, 1893–1907 CrossRef CAS.
- B. L. Hartwell, C. J. Pickens, M. Leon, L. Northrup, M. Christopher, J. D. Griffin, F. Martinez-Becerra and C. Berkland, J. Autoimmun., 2018, 93, 76–88 CrossRef CAS.
- R. A. Maldonado, R. A. LaMothe, J. D. Ferrari, A. H. Zhang, R. J. Rossi, P. N. Kolte, A. P. Griset, C. O'Neil, D. H. Altreuter, E. Browning, L. Johnston, O. C. Farokhzad, R. Langer, D. W. Scott, U. H. von Andrian and T. K. Kishimoto, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E156–E165 CrossRef CAS.
- R. A. LaMothe, P. N. Kolte, T. Vo, J. D. Ferrari, T. C. Gelsinger, J. Wong, V. T. Chan, S. Ahmed, A. Srinivasan, P. Deitemeyer, R. A. Maldonado and T. K. Kishimoto, Front. Immunol., 2018, 9, 281 CrossRef.
- A. H. Zhang, R. J. Rossi, J. Yoon, H. Wang and D. W. Scott, Cell. Immunol., 2016, 301, 74–81 CrossRef CAS.
- K. J. Peine, M. Guerau-de-Arellano, P. Lee, N. Kanthamneni, M. Severin, G. D. Probst, H. Peng, Y. Yang, Z. Vangundy, T. L. Papenfuss, A. E. Lovett-Racke, E. M. Bachelder and K. M. Ainslie, Mol. Pharm., 2014, 11, 828–835 CrossRef CAS.
- N. Chen, K. J. Peine, M. A. Collier, S. Gautam, K. A. Jablonski, M. Guerau-de-Arellano, K. M. Ainslie and E. M. Bachelder, Adv. Biosyst., 2017, 1, 1700022 CrossRef.
- N. Chen, C. J. Kroger, R. M. Tisch, E. M. Bachelder and K. M. Ainslie, Adv. Healthcare Mater., 2018, 7, 1800341 CrossRef.
- A. J. Giles, M. K. N. Hutchinson, H. M. Sonnemann, J. Jung, P. E. Fecci, N. M. Ratnam, W. Zhang, H. Song, R. Bailey and D. Davis, J. Immunother. Cancer, 2018, 6, 1–13 Search PubMed.
- L. H. Tostanoski, Y. C. Chiu, J. M. Gammon, T. Simon, J. I. Andorko, J. S. Bromberg and C. M. Jewell, Cell Rep., 2016, 16, 2940–2952 CrossRef CAS.
- L. Kappos, G. Comi, H. Panitch, J. Oger, J. Antel, P. Conlon, L. Steinman, A. Rae-Grant, J. Castaldo and N. Eckert, Nat. Med., 2000, 6, 1176–1182 CrossRef CAS.
- A. Lathuilière, N. Mach and B. L. Schneider, Int. J. Mol. Sci., 2015, 16, 10578–10600 CrossRef.
- M. E. Dudley and S. A. Rosenberg, Nat. Rev. Cancer, 2003, 3, 666–675 CrossRef CAS.
- S. A. Rosenberg and N. P. Restifo, Science, 2015, 348, 62–68 CrossRef CAS.
- S. A. Rosenberg, N. P. Restifo, J. C. Yang, R. A. Morgan and M. E. Dudley, Nat. Rev. Cancer, 2008, 8, 299–308 CrossRef CAS.
- Y. Honaker, N. Hubbard, Y. Xiang, L. Fisher, D. Hagin, K. Sommer, Y. Song, S. J. Yang, C. Lopez, T. Tappen, E. M. Dam, I. Khan, M. Hale, J. H. Buckner, A. M. Scharenberg, T. R. Torgerson and D. J. Rawlings, Sci. Transl. Med., 2020, 12, eaay6422 CrossRef CAS.
- F. Casiraghi, G. Remuzzi and N. Perico, Curr. Opin. Organ Transplant., 2014, 19, 47–53 CrossRef CAS.
- J. M. Ryan, F. P. Barry, J. M. Murphy and B. P. Mahon, J. Inflammation, 2005, 2, 8 CrossRef.
- H. Qi, G. Chen, Y. Huang, Z. Si and J. Li, J. Transl. Med., 2015, 13, 274 CrossRef.
- R. Ciccocioppo, A. Camarca, G. C. Cangemi, G. Radano, S. Vitale, E. Betti, D. Ferrari, L. Visai, E. Strada, C. Badulli, F. Locatelli, C. Klersy, C. Gianfrani and G. R. Corazza, Cytotherapy, 2014, 16, 1080–1091 CrossRef CAS.
- A. Bartholomew, C. Sturgeon, M. Siatskas, K. Ferrer, K. McIntosh, S. Patil, W. Hardy, S. Devine, D. Ucker, R. Deans, A. Moseley and R. Hoffman, Exp. Hematol., 2002, 30, 42–48 CrossRef.
- A. Y. Clark, K. E. Martin, J. R. García, C. T. Johnson, H. S. Theriault, W. M. Han, D. W. Zhou, E. A. Botchwey and A. J. García, Nat. Commun., 2020, 11, 114 CrossRef CAS.
- E. Y. Yang, J. P. Kronenfeld, K. M. Gattás-Asfura, A. L. Bayer and C. L. Stabler, Biomaterials, 2015, 67, 20–31 CrossRef CAS.
- S. D. Miller, R. P. Wetzig and H. N. Claman, J. Exp. Med., 1979, 149, 758–773 CrossRef CAS.
- C. B. Smarr, C. L. Hsu, A. J. Byrne, S. D. Miller and P. J. Bryce, J. Immunol., 2011, 187, 5090–5098 CrossRef CAS.
- T. Kheradmand, S. Wang, J. Bryant, J. J. Tasch, N. Lerret, K. L. Pothoven, J. L. Houlihan, S. D. Miller, Z. J. Zhang and X. Luo, J. Immunol., 2012, 189, 804–812 CrossRef CAS.
- X. Luo, K. L. Pothoven, D. McCarthy, M. DeGutes, A. Martin, D. R. Getts, G. Xia, J. He, X. Zhang, D. B. Kaufman and S. D. Miller, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14527–14532 CrossRef CAS.
- T. Kheradmand, S. Wang, R. F. Gibly, X. Zhang, S. Holland, J. Tasch, J. G. Graham, D. B. Kaufman, S. D. Miller, L. D. Shea and X. Luo, Biomaterials, 2011, 32, 4517–4524 CrossRef CAS.
- A. Lutterotti, S. Yousef, A. Sputtek, K. H. Stürner, J. P. Stellmann, P. Breiden, S. Reinhardt, C. Schulze, M. Bester, C. Heesen, S. Schippling, S. D. Miller, M. Sospedra and R. Martin, Sci. Transl. Med., 2013, 5, 188ra75–188ra75 Search PubMed.
- S. Sriram, G. Schwartz and L. Steinman, Cell. Immunol., 1983, 75, 378–382 CrossRef CAS.
- X. M. Su and S. Sriram, J. Neuroimmunol., 1991, 34, 181–190 CrossRef CAS.
- M. K. Kennedy, L. J. Tan, M. C. Dal Canto, V. K. Tuohy, Z. J. Lu, J. L. Trotter and S. D. Miller, J. Immunol., 1990, 144, 909–915 CAS.
- M. K. Kennedy, L. J. Tan, M. C. Dal Canto and S. D. Miller, J. Immunol., 1990, 145, 117–126 CAS.
- A. Abdel-Gadir, A. H. Massoud and T. A. Chatila, F1000Research, 2018, 7, 38 Search PubMed.
- L. J. Tan, M. K. Kennedy, M. C. Dal Canto and S. D. Miller, J. Immunol., 1991, 147, 1797–1802 CAS.
- C. L. Vanderlugt, K. L. Neville, K. M. Nikcevich, T. N. Eagar, J. A. Bluestone and S. D. Miller, J. Immunol., 2000, 164, 670–678 CrossRef CAS.
- C. E. Smith and S. D. Miller, J. Autoimmun., 2006, 27, 218–231 CrossRef CAS.
- I. Zubizarreta, G. Flórez-Grau, G. Vila, R. Cabezón, C. España, M. Andorra, A. Saiz, S. Llufriu, M. Sepulveda, N. Sola-Valls, E. H. Martinez-Lapiscina, I. Pulido-Valdeolivas, B. Casanova, M. Martinez Gines, N. Tellez, C. Oreja-Guevara, M. Español, E. Trias, J. Cid, M. Juan, M. Lozano, Y. Blanco, L. Steinman, D. Benitez-Ribas and P. Villoslada, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8463–8470 CrossRef CAS.
- N. Pishesha, A. M. Bilate, M. C. Wibowo, N. J. Huang, Z. Li, R. Deshycka, D. Bousbaine, H. Li, H. C. Patterson, S. K. Dougan, T. Maruyama, H. F. Lodish and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3157–3162 CrossRef CAS.
- N. Askenasy, E. S. Yolcu, Z. Wang and H. Shirwan, Circulation, 2003, 107, 1525–1531 CrossRef CAS.
- H. Zhao, K. Woodward, H. Shirwan and E. Yolcu, Transplantation, 2012, 94, 483 CrossRef.
- H. Zhao, K. B. Woodward, H. Shirwan, O. Grimany-Nuno and E. S. Yolcu, Transplant. Proc., 2013, 45, 1805–1807 CrossRef CAS.
- E. S. Yolcu, N. Askenasy, N. P. Singh, S. E. L. Cherradi and H. Shirwan, Immunity, 2002, 17, 795–808 CrossRef CAS.
- E. S. Yolcu, H. Zhao, L. Bandura-Morgan, C. Lacelle, K. B. Woodward, N. Askenasy and H. Shirwan, J. Immunol., 2011, 187, 5901–5909 CrossRef CAS.
- K. B. Woodward, H. Zhao, P. Shrestha, L. Batra, M. Tan, O. Grimany-Nuno, L. Bandura-Morgan, N. Askenasy, H. Shirwan and E. S. Yolcu, Am. J. Transplant., 2020, 20, 1285–1295 CrossRef CAS.
- P. Shrestha, L. Batra, M. Tariq Malik, M. Tan, E. S. Yolcu and H. Shirwan, Am. J. Transplant., 2020, 20 DOI:10.1111/ajt.15958.
- L. Batra, P. Shrestha, H. Zhao, K. B. Woodward, A. Togay, M. Tan, O. Grimany-Nuno, M. T. Malik, M. M. Coronel, A. J. García, H. Shirwan and E. S. Yolcu, J. Immunol., 2020, 204, 2840–2851 CrossRef CAS.
- L. Batra, P. Shrestha, E. S. Yolcu, H. Zhao, W. S. Bowen, K. B. Woodward, M. M. Coronel, M. Tan, A. J. García and H. Shirwan, Transplantation, 2018, 102, S455 CrossRef.
- N. Navarro-Alvarez and Y. G. Yang, Cell. Mol. Immunol., 2011, 8, 285–288 CrossRef CAS.
- R. M. Hernández, G. Orive, A. Murua and J. L. Pedraz, Adv. Drug Delivery Rev., 2010, 62, 711–730 CrossRef.
- C. Wang, R. R. Varshney and D. A. Wang, Adv. Drug Delivery Rev., 2010, 62, 699–710 CrossRef CAS.
- S. M. Willerth and S. E. Sakiyama-Elbert, StemJournal, 2019, 1, 1–25 Search PubMed.
- S. Sakaguchi, N. Sakaguchi, M. Asano, M. Itoh and M. Toda, J. Immunol., 1995, 155, 1151–1164 CAS.
- N. Safinia, C. Scotta, T. Vaikunthanathan, R. I. Lechler and G. Lombardi, Front. Immunol., 2015, 6, 438 Search PubMed.
- B. D. Singer, L. S. King and F. R. D'Alessio, Front. Immunol., 2014, 5, 46 Search PubMed.
- J. A. Bluestone, J. H. Buckner, M. Fitch, S. E. Gitelman, S. Gupta, M. K. Hellerstein, K. C. Herold, A. Lares, M. R. Lee, K. Li, W. Liu, S. A. Long, L. M. Masiello, V. Nguyen, A. L. Putnam, M. Rieck, P. Sayre and Q. Tang, Sci. Transl. Med., 2015, 7, 315ra189 CrossRef.
- C. G. Brunstein, J. S. Miller, Q. Cao, D. H. McKenna, K. L. Hippen, J. Curtsinger, T. DeFor, B. L. Levine, C. H. June, P. Rubinstein, P. B. McGlave, B. R. Blazar and J. E. Wagner, Blood, 2011, 117, 1061–1070 CrossRef CAS.
- K. C. Santos Roballo, S. Dhungana, Z. Jiang, J. Oakey and J. S. Bushman, Biomaterials, 2019, 209, 1–9 CrossRef CAS.
- N. Marek, A. Krzystyniak, I. Ergenc, O. Cochet, R. Misawa, L. J. Wang, K. Gołąb, X. Wang, G. Kilimnik, M. Hara, S. Kizilel, P. Trzonkowski, J. M. Millis and P. Witkowski, Ann. Surg., 2011, 254, 512–519 CrossRef.
- J. G. Graham, X. Zhang, A. Goodman, K. Pothoven, J. Houlihan, S. Wang, R. M. Gower, X. Luo and L. D. Shea, Tissue Eng., Part A, 2013, 19, 1465–1475 CrossRef CAS.
- J. A. Ankrum, J. F. Ong and J. M. Karp, Nat. Biotechnol., 2014, 32, 252 CrossRef CAS.
- M. B. Murphy, K. Moncivais and A. I. Caplan, Exp. Mol. Med., 2013, 45, e54 CrossRef.
- K. Lee, Y. Xue, J. Lee, H. J. Kim, Y. Liu, P. Tebon, E. Sarikhani, W. Sun, S. Zhang, R. Haghniaz, B. Çelebi-Saltik, X. Zhou, S. Ostrovidov, S. Ahadian, N. Ashammakhi, M. R. Dokmeci and A. Khademhosseini, Adv. Funct. Mater., 2020, 30, 2000086 CrossRef CAS.
- X. Zhang, K. Yamaoka, K. Sonomoto, H. Kaneko, M. Satake, Y. Yamamoto, M. Kondo, J. Zhao, I. Miyagawa and K. Yamagata, PLoS One, 2014, 9, e114621 CrossRef.
- O. Levy, R. Kuai, E. M. Siren, D. Bhere, Y. Milton, N. Nissar, M. De Biasio, M. Heinelt, B. Reeve and R. Abdi, Sci. Adv., 2020, 6, eaba6884 CrossRef CAS.
- K. Sadtler, B. W. Allen, K. Estrellas, F. Housseau, D. M. Pardoll and J. H. Elisseeff, Tissue Eng., Part A, 2016, 23, 1044–1053 CrossRef.
- K. Sadtler, A. Singh, M. T. Wolf, X. Wang, D. M. Pardoll and J. H. Elisseeff, Nat. Rev. Mater., 2016, 1, 1–17 CrossRef.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |