
Engineering a macroporous fibrin-based sequential interpenetrating polymer network for dermal tissue engineering†
Olfat
Gsib
a,
Loek J.
Eggermont
b,
Christophe
Egles
a and
Sidi A.
Bencherif
 *abcd
*abcd
aLaboratoire de BioMécanique et BioIngénierie (BMBI), UMR CNRS 7388, Sorbonne Universités, Université de Technologie of Compiègne (UTC), Compiègne, France. E-mail: s.bencherif@northeastern.edu
bDepartments of Chemical Engineering and Bioengineering, Northeastern University, Boston, MA, USA
cDepartment of Bioengineering, Northeastern University, Boston, MA, USA
dHarvard John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA, USA
First published on 12th October 2020
Abstract
The success of skin tissue engineering for deep wound healing relies predominantly on the design of innovative and effective biomaterials. This study reports the synthesis and characterization of a new type of naturally-derived and macroporous interpenetrating polymer network (IPN) for skin repair. These biomaterials consist of a biologically active fibrous fibrin network polymerized within a mechanically robust and macroporous construct made of polyethylene glycol and biodegradable serum albumin (PEGDM-co-SAM). First, mesoporous PEGDM-co-SAM hydrogels were synthesized and subjected to cryotreatment to introduce an interconnected macroporous network. Subsequently, fibrin precursors were incorporated within the cryotreated PEG-based network and then allowed to spontaneously polymerize and form a sequential IPN. Rheological measurements indicated that fibrin-based sequential IPN hydrogels exhibited improved and tunable mechanical properties when compared to fibrin hydrogels alone. In vitro data showed that human dermal fibroblasts adhere, infiltrate and proliferate within the IPN constructs, and were able to secrete endogenous extracellular matrix proteins, namely collagen I and fibronectin. Furthermore, a preclinical study in mice demonstrated that IPNs were stable over 1-month following subcutaneous implantation, induced a minimal host inflammatory response, and displayed a substantial cellular infiltration and tissue remodeling within the constructs. Collectively, these data suggest that macroporous and mechanically reinforced fibrin-based sequential IPN hydrogels are promising three-dimensional platforms for dermal tissue regeneration.
Introduction
Over the years, considerable evidence has suggested that various extracellular matrix (ECM) components play a critical role in wound healing.1–5 Since collagen is the most abundant protein in the dermis layer of the skin, collagen-based scaffolds were among the first artificial ECMs to be developed for dermal tissue regeneration.6,7 However, their clinical applications have been hindered by several drawbacks such as poor structural integrity and unsatisfactory esthetic outcomes post-implantation.6,8,9 Other biopolymers including chitosan,3,10 elastin,8,9,11 hyaluronic acid12 and gelatin1,13 have been investigated for skin scaffold design as well. Yet, these biomaterials did not properly mimic the provisional extracellular matrix formed during the healing process, thereby greatly contributing to irregular scarring after their implantation.6,8,9,14Fibrin, a naturally-occurring biopolymer involved in wound healing, has been widely used in tissue engineering due to its bioactivity, biocompatibility, biodegradability and facile processability.14 Fibrin-based matrices serve as temporary scaffolds during wound healing in which cells such as fibroblasts adhere, migrate, proliferate and gradually synthesized de novo ECM.14 In addition to its healing properties, fibrin provides the advantage of having its precursors, fibrinogen, and thrombin, easily extractable from blood samples of patients making autologous treatment possible.14–16 Current fibrin-based biomaterials vary from sheets17 to sealants18 and hydrogels.19–23 Soft, bioactive and tissue-like fibrin-based hydrogels are appealing due to their high water content, making them resemble native soft tissues.24 Additionally, when fibrin hydrogels are synthesized under physiological conditions, they display biological functionalities similar to those found in the native dermal microenvironment.14,15
However, the weak mechanical properties of fibrin hydrogels usually prevent their application in load-bearing situations.14,25 To overcome this limitation, several strategies have been implemented to improve their physical properties. For instance, increasing the concentration of fibrinogen led to stiffer hydrogels,26 but resulted in microstructures that were too dense for three-dimensional (3D) cell culture.27,28 Conjugating fibrinogen with synthetic polymers such as polyethylene glycol (PEGylation) was also tested to enhance the mechanical stability of fibrin hydrogels. However, fibrin-based hydrogels made with PEGylated fibrinogen did not perform as well as those made with unmodified fibrinogen.29
Alternatively, interpenetrating polymer networks (IPNs) provide great means to structurally reinforce naturally-derived hydrogels.1,30–32 An IPN is a construct comprising of at least two polymer networks that are intertwined on the molecular scale without covalent bonds between them.33,34 Various synthetic routes have been investigated to fabricate IPNs. IPNs are either (i) fabricated with a simultaneous pathway by creating all polymer chains at the same time by independent, non-interfering routes26,34 or (ii) with a sequential pathway, in which the secondary network is created around the first one, once fully synthesized.33,35
IPNs combining fibrin fibers entangled with other biopolymers exhibit reinforced mechanical strengths when compared with each polymer network individually.30,36,37 Incorporating synthetic polymers such as polyethylene glycol (PEO or PEG)26 and polyvinyl alcohol (PVA)28 could further improve their physical properties. While these semi-synthetic IPNs exhibited suitable mechanical and biological characteristics, they did not provide suitable degradable networks.26,28 Serum albumin (SA), a naturally-derived and degradable protein, has been incorporated into synthetic IPNs to enable enzymatic biodegradation.38 Yet, these IPNs did not exhibit large and interconnected pores which are of critical importance for tissue engineering applications.39,40 Ideally, IPNs should display interconnected macroporous architectures to ensure neovascularization,41 cellular infiltration and organization,42,43 adequate diffusion of nutrients to cells within the construct as well as de novo tissue synthesis and remodeling.39,40,42,43
To address these limitations, we recently engineered and characterized a new type of macroporous sequential IPN hydrogels combining the intrinsic biological properties of fibrin with the remarkable mechanical features of PEG. First, methacrylated SA (SAM) was copolymerized with PEG dimethacrylate (PEGDM) to form enzymatically degradable PEGDM-co-SAM hydrogels. Next, these hydrogels were subjected to several freeze–thaw cycles (i.e., cryotreatment) to create interconnected macroporous networks. Lastly, cryotreated PEGDM-co-SAM hydrogels (1st intertwined network) were swollen into aqueous solutions (water or HEPES buffer) containing fibrin precursors (fibrinogen and thrombin) which were sequentially polymerized to form a 2nd intertwined network, giving rise to sequential IPNs. The physico-chemical properties of IPNs, consisting of cryotreated PEGDM-co-SAM networks entangled with fibrin fibers, were thoroughly characterized. Subsequently, their biological properties were assessed using human dermal fibroblasts (HDFs) from neonatal foreskin. Lastly, preliminary in vivo studies were performed to assess their biocompatibility, biointegration, and biostability in mice.
Experimental
Materials
Polyethylene glycol (PEG, 4k) (MM = 4000 g mol−1), methacrylic anhydride (MA), diethyl ether, dichloromethane, triethylamine (TEA), ammonium persulfate (APS), N,N,N′,N′-tetramethylethylenediamine (TEMED), sodium chloride, calcium chloride, methacrylic acid N-hydroxysuccinimide ester (NHSM, purity ∼98%), albumin from bovine serum (purity 98%), sodium chloride, propidium iodide, Hoechst 33342, anti-fibronectin antibodies produced in rabbit, acetone were purchased from Sigma-Aldrich. Deuterium oxide and NMR tubes were purchased from Eurisotop. CellTiter 96® AQueous one solution cell proliferation assay (MTS) was obtained from Promega. Penicillin–streptomycin, L-glutamine 1 mM, trypsin–EDTA (0.05%), Click-iT EdU Alexa Fluor 488 imaging kit, calcein AM, goat anti-rabbit IgG secondary antibodies coupled to Alexa Fluor 633 conjugates and goat anti-mouse IgG secondary antibodies coupled to Alexa fluor 488 conjugates were purchased from Life Technologies. Dulbecco's modified eagle medium (DMEM), serum HyClone and six mm biopsy punches were obtained from Dutscher. HDFs from neonatal foreskin were purchased from Cascade Biologics™. Rabbit fluorescein isothiocyanate (FITC)-labelled anti-fibrinogen antibodies were obtained from Dako, North America, Inc. Thrombin from bovine plasma was purchased from Fisher scientific and bovine fibrinogen was obtained from Calbiochem. NHS-rhodamine (5/6-carboxy-tetramethyl-rhodamine succinimidyl ester) mixed isomer was purchased from ThermoFisher. HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, purity 99%) was purchased from Acros Organics. Boric acid and mouse monoclonal, sterile (50 ml) 0.22 μm disposable vacuum filtration system Steriflip® and anti-collagen type I antibodies were purchased from Merck Millipore. Phalloidin fluor 647 reagent was obtained from Abcam. Paraformaldehyde (PFA) 16% solution was purchased from Oxford Instruments. Formaldehyde 4% buffered (pink) Q Path®, saffron alcoholic solution Q Path® and optimal cutting temperature (OCT) compound were obtained from VWR. For animal studies, 5-week old nude mice (NMRI-Foxn1/foxn1nu, male) were purchased from Janvier Labs. Biological implants CELLIS® composed of acellular porcine collagen dermis were obtained from Meccellis Biotech. POLIGLECAPRONE 25 sutures were purchased from Vetsuture®.Synthesis and characterization of PEGDM
PEGDM was prepared from 4K PEG and MA. PEG (20 g, 0.005 mol), MA (3.86 g, 0.025 mol) and TEA (0.8 mL) were reacted in dichloromethane (60 mL) for 2 days at room temperature. The solution was then precipitated into diethyl ether and the product was subsequently filtered and dried in a vacuum oven for 5 days at RT. 1H nuclear magnetic resonance (NMR) spectra (270 MHz) were taken on an NMR Avance DSX 400 (Bruker, Germany) to evaluate the degree of functionalization of PEGDM (Fig. S1†). Deuterated oxide (D2O) was used as a solvent and the polymer concentrations were 2.0% by mass fraction. All spectra were run at RT, 20 Hz sample spinning, 30° tip angle for the observation pulse, and a 10 s recycle delay, for 64 scans.Synthesis and characterization of SAM
Bovine SA was functionalized with methacrylate residues following a previously published procedure.15,38 Briefly, 4% w/v bovine SA was solubilized in 0.25 M boric acid buffer at pH 7.4 and then incubated at 37 °C until complete dissolution. Next, NHSM was dissolved in acetone and added dropwise to the SA solution. The reaction took place under stirring overnight at RT. The resulting SAM was then purified by dialysis against 0.01 M HEPES at pH 7.4. Dialysis baths were replaced 3 times (after 2, 6 and 24 h). Next, purified SAM was lyophilized and stored at −80 °C. Before use, SAM was weighed, dissolved in water (200 mg ml−1) and sterile filtered (0.22 μm). A TNBS assay was performed to evaluate the degree of functionalization of SAM (Fig. S2†).Fabrication of fibrin/PEGDM-co-SAM IPN hydrogels
A schematic of the hydrogel formation is presented in Fig. 1. Different formulations of PEGDM-co-SAM hydrogels were prepared by mixing PEGDM at various concentrations with SAM in deionized water. The concentrations of (5% w/v) of PEGDM and (5% v/v) of SAM were identified as described by others,28,38 using similar formulations. Additionally, a higher concentration of PEGDM (15% v/v) was also tested in order to obtain hydrogels with higher stiffnesses.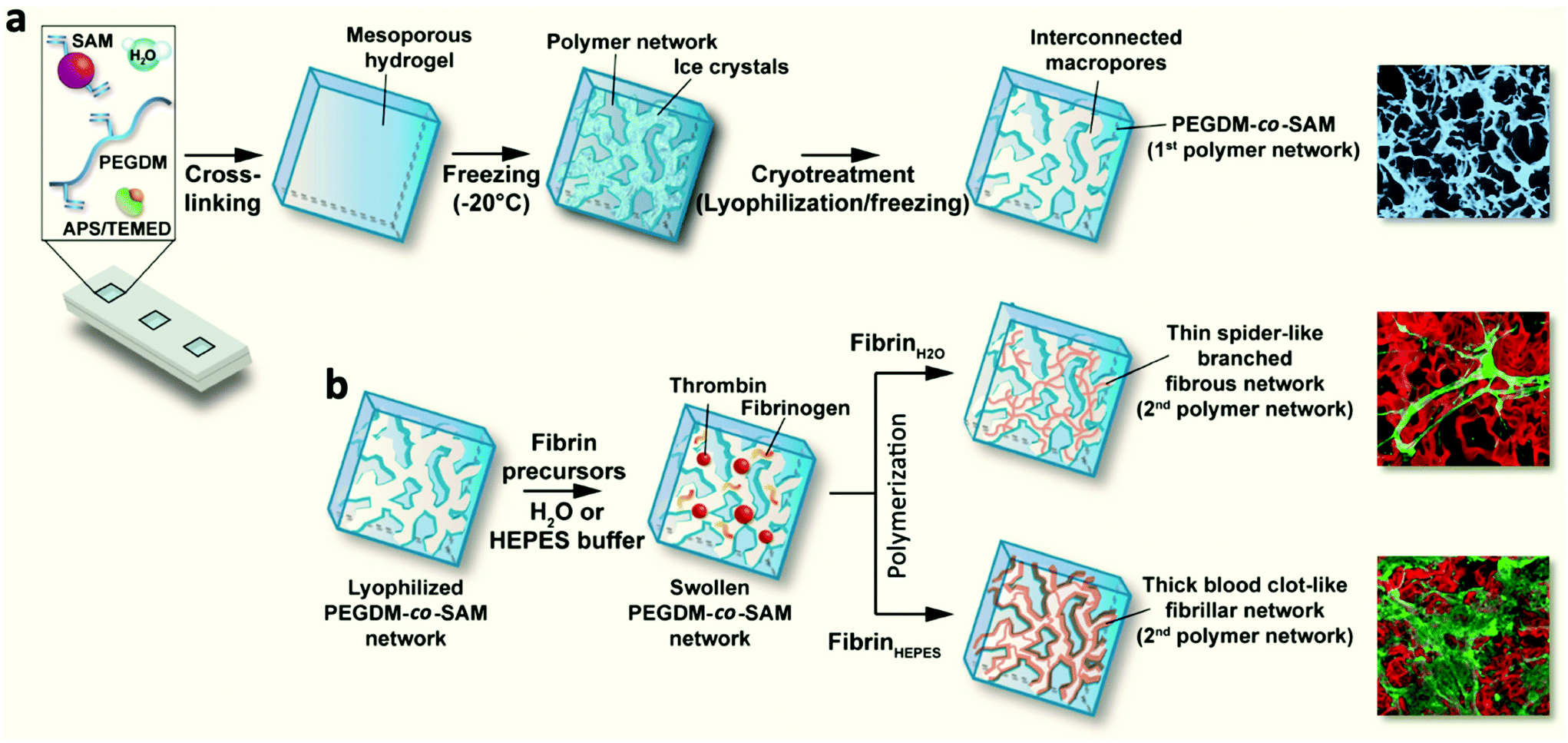 | ||
| Fig. 1 Schematic depicting the fabrication process of macroporous fibrin/PEGDM-co-SAM sequential IPNs. (a) Fabrication of the first crosslinked PEGDM-co-SAM network. PEGDM and SAM at various concentrations were copolymerized in water using an APS/TEMED initiator system to fabricate mesoporous PEGDM-co-SAM hydrogels. PEGDM-co-SAM hydrogels were subsequently frozen at −20 °C and subjected to cryotreatment (i.e., 3 freeze–thaw cycles) to create an interconnected macroporous structure. Pseudocolored (blue) SEM image (right) depicting the macroporous architecture of a cryotreated PEGDM-co-SAM hydrogel. (b) Sequential polymerization of the second fibrin network. Cryotreated and lyophilized PEGDM-co-SAM scaffolds were swollen with aqueous solvents (HEPES buffer or water) containing fibrin precursors (fibrinogen and thrombin) and polymerized under physiological conditions to form sequential IPNs. Confocal images (right) depicting fibrin (green) and PEGDM-co-SAM (red) sequential IPNs. | ||
APS and TEMED (Sigma-Aldrich) solutions were added to the pre-gel solutions at the following concentrations: 0.4% and 1.6% v/v for PEGDM5SAM5 and 0.1% and 0.4% v/v for PEGDM15SAM5, respectively. Immediately after mixing, the solutions were quickly vortexed and then transferred into Teflon molds. The formed hydrogels were then swollen in deionized water, stored at −20 °C for 2 h and freeze-dried. This cryotreatment process was repeated three times. Next, a total of two fibrin formulations in water (0.5 and 1% w/v) were formed by mixing bovine fibrinogen with thrombin (0.2 U ml−1) in deionized water. These fibrin formulations were referenced as Fb(0.5%)H2O and Fb(1%)H2O, respectively. Fb(0.5%)HEPES networks were also formed by mixing the fibrin precursors in a HEPES buffer, pH 7.4 (0.01 M HEPES, 0.15 M NaCl, and 0.02 M CaCl2). Next, freeze-dried PEGDM-co-SAM constructs were rehydrated with an aqueous solvent containing the fibrin precursors and allowed to sequentially polymerize and form macroporous fibrin/PEGDM-co-SAM IPNs.
Evaluation of the microstructure
Environmental scanning electron microscopy (ESEM) (XL 30-ESEM® FEG, Philips, Eindhoven, The Netherlands) was used to image the microstructural features of PEGDM-co-SAM and fibrin/PEGDM-co-SAM gel samples (Fig. 2). Pore diameters were quantified from the collected ESEM images. Diameters of the longest axes in each of 5 pores per image for a total of 10 images per sample were quantified using Image J software. Confocal laser scanning microscopy (CLSM) (Zeiss LSM 410 invert, Oberkochen, Germany) was used to image the fibrin and PEG networks. PEGDM-co-SAM network was labelled with NHS-rhodamine and fibrin was stained with FITC-labelled anti-fibrinogen antibodies.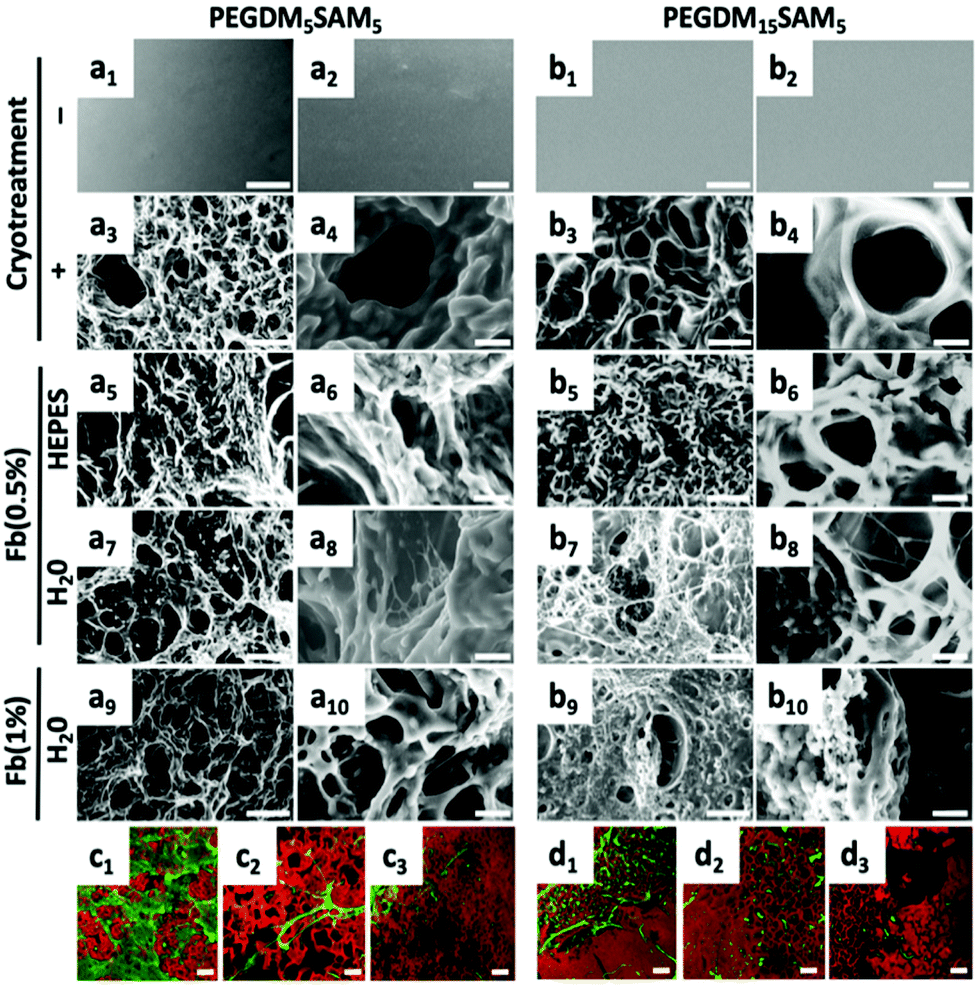 | ||
| Fig. 2 ESEM and confocal images of pre- and post-cryotreated PEGDM-co-SAM and fibrin/PEGDM-co-SAM sequential IPN hydrogels. PEGDM5SAM5 (a) and PEGDM15SAM5 (b) hydrogels were imaged with ESEM pre- (a1, a2, b1, b2) and post- (a3, a4, b3, b4) cryotreatment. fibrin/PEGDM-co-SAM sequential IPN hydrogels were also imaged across the three investigated fibrin formulations: Fb(0.5%)HEPES (a5, a6, b5, b6), Fb(0.5%)H2O (a7, a8, b7, b8) and Fb(1%)H2O (a9, a10, b9, b10). Scale bars = 100 μm (1, 3, 5, 7, 9) and 20 μm (2, 4, 6, 8 and 10). a2, a4, a6, a8, a10 and b2, b4, b6, b8, b10 are zoomed-in views of images a1, a3, a5, a7, a9 and b1, b3, b5, b7, b9, respectively. Z-Stack confocal images of PEGDM5SAM5 (c) or PEGDM15SAM5 (d) network stained in red with rhodamine-labelled SAM. The sequential fibrin network was labelled in green with FITC-labelled anti-fibrinogen antibodies for each formulation investigated: Fb(0.5%)HEPES (1) (c1 and d1), Fb(0.5%)H2O (c2 and d2), and Fb(1%)H2O (c3 and d3). Scale bars = 100 μm (c and d). | ||
Swelling ratio measurements
Hydrogels were swollen up to 5 h in phosphate buffered saline (PBS) at 37 °C to reach equilibrium and their weights recorded. The hydrogels were then washed several times in deionized water. The gel samples were subsequently dried, and their weights recorded. The mass swelling ratio (QM) was calculated based on eqn (1):| QM = Ws/Wd | (1) |
 | ||
Fig. 3 Swelling characteristics of the various (a) fibrin formulations and (b and c) cryotreated PEGDM-co-SAM: swelling kinetics for (a1) fibrin hydrogels and across fibrin-free and fibrin-containing PEGDM-co-SAM hydrogels: (b1) PEGDM5SAM5 and (c1) PEGDM15SAM5. Swelling mass ratio (QM) at equilibrium for (a2) fibrin hydrogels and across fibrin-free and fibrin-containing PEGDM-co-SAM hydrogels: (b2) PEGDM5SAM5 and (c2) PEGDM15SAM5. The swelling ratios QM at equilibrium measured on the different fibrin-containing PEGDM-co-SAM hydrogels were compared to those of fibrin-free cryotreated PEGDM-co-SAM hydrogels and to each other. Values represent mean ± SD (![[n with combining low line]](https://www.rsc.org/images/entities/i_char_006e_0332.gif) ≥ 3). Data were analyzed using one-way ANOVA test. *p < 0.05, **p < 0.01, and ***p < 0.001. ≥ 3). Data were analyzed using one-way ANOVA test. *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
Mechanical measurements
Rheological measurements of hydrogels (sample dimensions: 50 mm in diameter and 1 mm in height) were performed using an Anton Paar Physica MCR 301 rheometer with a plate–plate geometry (diameter: 50 mm). We first performed strain and frequency sweeps on PEGDM-co-SAM and fibrin hydrogels to select the appropriate strain amplitude (1%) and frequency (1 Hz) (Fig. S3 and S4†). Time sweeps were then performed to determine the gelation point (i.e., the time when storage (G′) and loss (G′′) moduli crossover) for each formulation and the equilibrium storage moduli (i.e., when G′ plateaued) for the different gel formulations either at RT (PEGDM-co-SAM) or at 37 °C (fibrin) (Fig. S5†). G′ and G′′ were next measured on lyophilized PEGDM-co-SAM constructs and fibrin/PEGDM-co-SAM IPN hydrogels (Fig. 4). The mechanical properties of fibrin/PEGDM-co-SAM IPN hydrogels were then compared to those of cryotreated PEGDM-co-SAM hydrogels and to each other.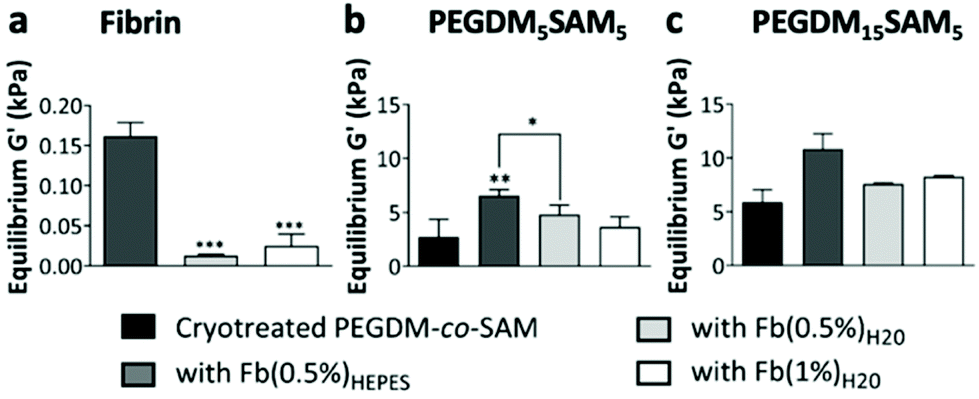 | ||
| Fig. 4 Elastic moduli at equilibrium for across the various (a) fibrin formulations and for (b and c) cryotreated PEGDM-co-SAM: (b) PEGDM5SAM5 and (c) PEGDM15SAM5 and their corresponding fibrin/PEGDM-co-SAM sequential IPN hydrogels when cryotreated PEGDM-co-SAM were sequentially polymerized with fibrin. The elastic moduli G′ at equilibrium for the different fibrin/PEGDM-co-SAM sequential IPN hydrogels were compared to those of cryotreated PEGDM-co-SAM hydrogels and to each other. Values represent mean ± SD (n ≥ 3). Data were analyzed using one-way ANOVA test. *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
Biological characterization of cell-laden fibrin/PEGDM-co-SAM IPN hydrogels
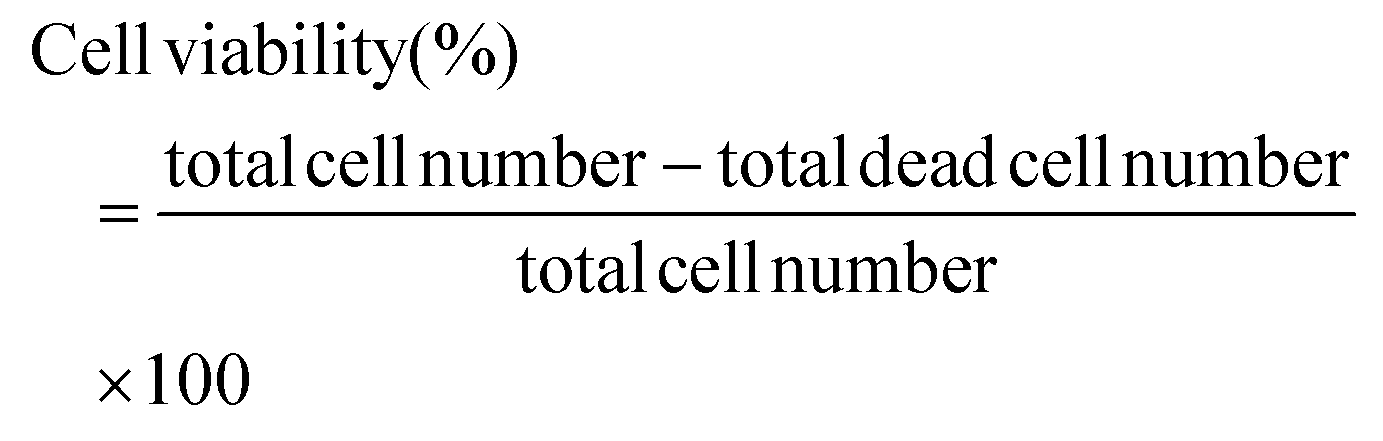 | (2) |
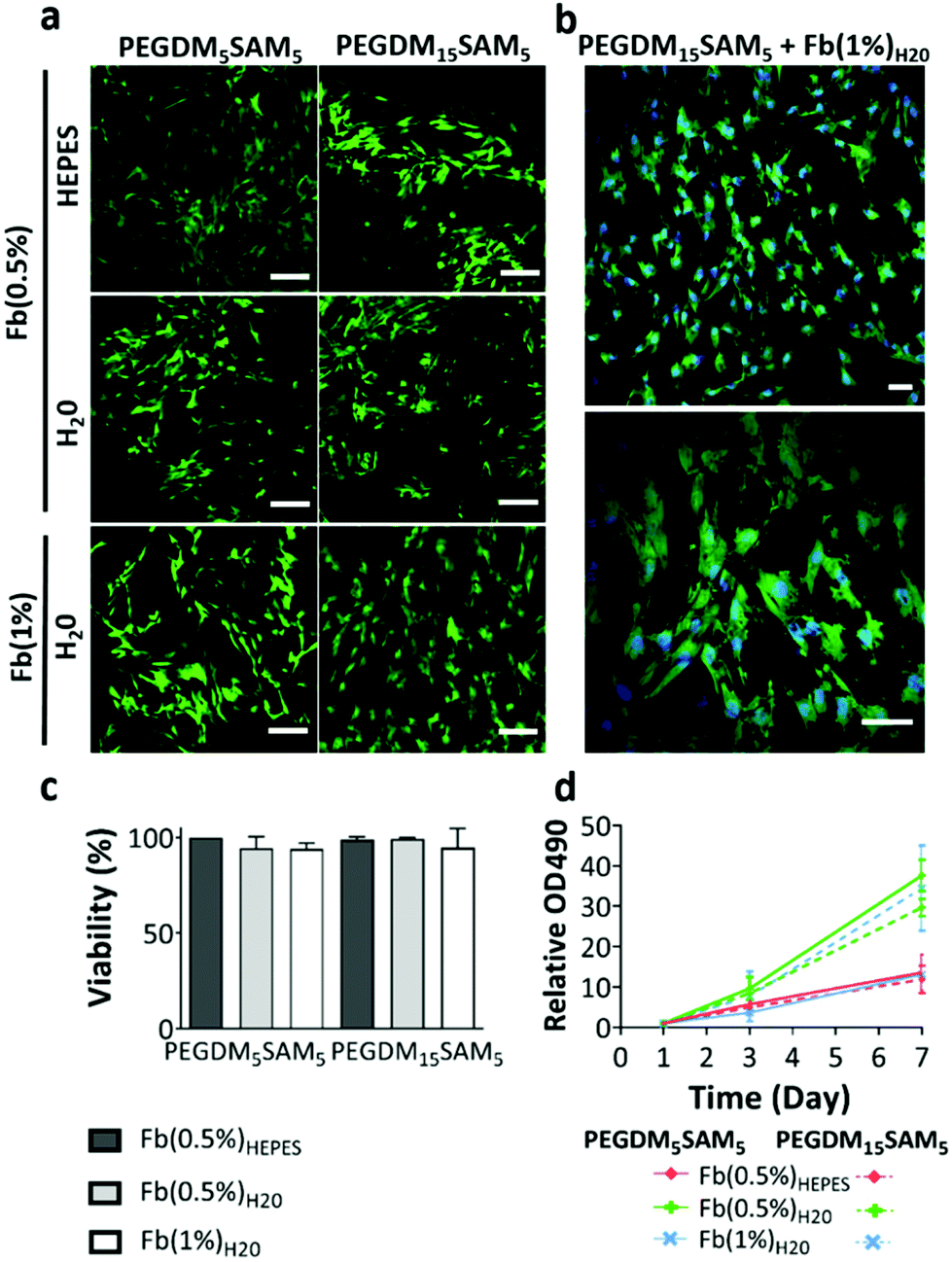 | ||
| Fig. 5 Evaluation of cell viability and metabolic activity of HDFs in fibrin/PEGDM-co-SAM sequential IPN hydrogels: (a) merged confocal microscopy images of HDFs cultured in IPNs after 2 d depicting live (green) and dead (red) cells. (b) Zoomed in images of HDF-laden IPN hydrogels made with Fb(1%)H2O showing cell nuclei (blue), live (green) and dead (red) cells. Scale bars = 100 μm (a) and 50 μm (b). (c) Cell viability (%) across fibrin/PEGDM-co-SAM sequential IPN hydrogels: fibrin/PEGDM5SAM5 (left) and fibrin/PEGDM15SAM5 (right). (d) Metabolic activity of HDFs in IPN hydrogels after 1, 3 and 7 d of culture. (c and d) Values represent mean ± SD (n ≥ 3). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 ratio and the samples (2 × 104 HDFs per construct) were then incubated for 2 h. The absorbance was measured at 490 nm with a microplate reader and data normalized to the day 1 absorbance (Fig. 5d).
:100) for 3 h. The samples were then imaged using a CLSM (Zeiss LSM 410 invert, Oberkochen, Germany). High-resolution image stacks were collected with 50 μm separation between slices (z-stacks) (Fig. 6, 7 and Fig. S6†).
:6 ratio and the samples (2 × 104 HDFs per construct) were then incubated for 2 h. The absorbance was measured at 490 nm with a microplate reader and data normalized to the day 1 absorbance (Fig. 5d).
:100) for 3 h. The samples were then imaged using a CLSM (Zeiss LSM 410 invert, Oberkochen, Germany). High-resolution image stacks were collected with 50 μm separation between slices (z-stacks) (Fig. 6, 7 and Fig. S6†).
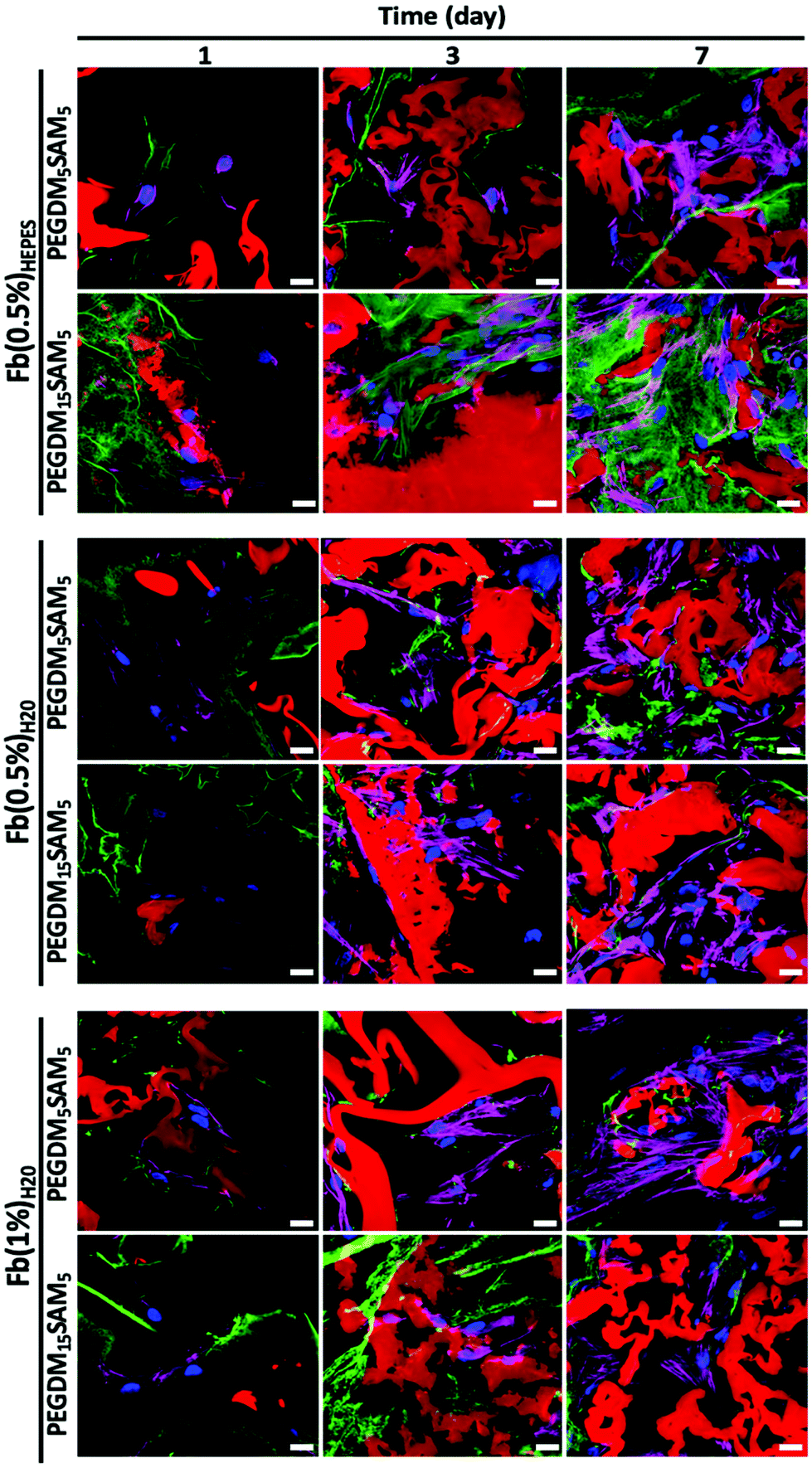 | ||
| Fig. 6 Cell morphology of HDFs in fibrin/PEGDM-co-SAM sequential IPN hydrogels. HDFs were cultured in various fibrin/PEGDM-co-SAM IPN constructs: fibrin/PEGDM5SAM5 and fibrin/PEGDM15SAM5 fabricated with different fibrin formulations: Fb(0.5%)HEPES, Fb(0.5%)H2O or Fb(1%)H2O. Cell morphology was analyzed by confocal microscopy after 1, 3, and 7 d of culture depicting F-actin (dark pink), nuclei (blue), fibrin (green) and PEGDM-co-SAM (red). Scale bar = 20 μm. | ||
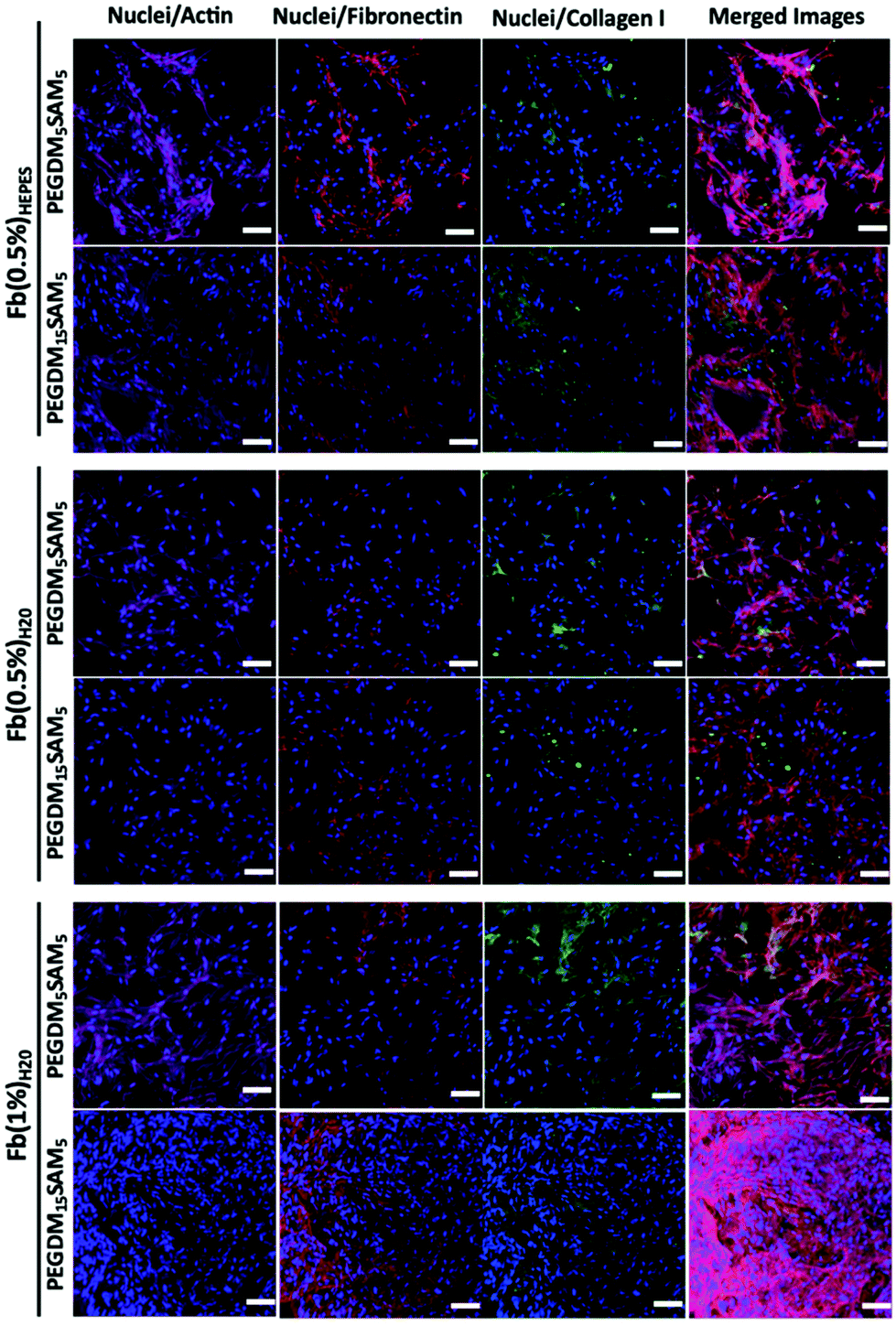 | ||
| Fig. 7 Evaluation of HDF-derived extracellular matrix deposition on fibrin/PEGDM-co-SAM sequential IPN hydrogels. Secretion of two endogenous extracellular matrix proteins (collagen I and fibronectin) from HFD after 1-week culture in various IPN constructs: fibrin/PEGDM5SAM5 and fibrin/PEGDM15SAM5 fabricated with different fibrin formulations: Fb(0.5%)HEPES, Fb(0.5%)H2O or Fb(1%)H2O. Samples were stained and analyzed by confocal microscopy: F-actin (dark pink), nuclei (blue), fibronectin (red), and collagen I (green). Scale bar = 100 μm. | ||
Mice were first anesthetized with 3.5% isoflurane and then the scaffolds (6 mm in diameter and 1 mm in height) were surgically implanted subcutaneously and sutured on both sides of the dorsal midline. Animals were sacrificed at week 4 following implantation. The scaffolds and the surrounding tissues were explanted and fixed in 4% formalin for 24 h. The biopsy specimens were then dehydrated using graded ethanol 70–96–100%, embedded in paraffin, sectioned at 10 μm, and finally stained with hematoxylin eosin (H&E) or hematoxylin eosin saffron (H&ES) (Fig. 8).
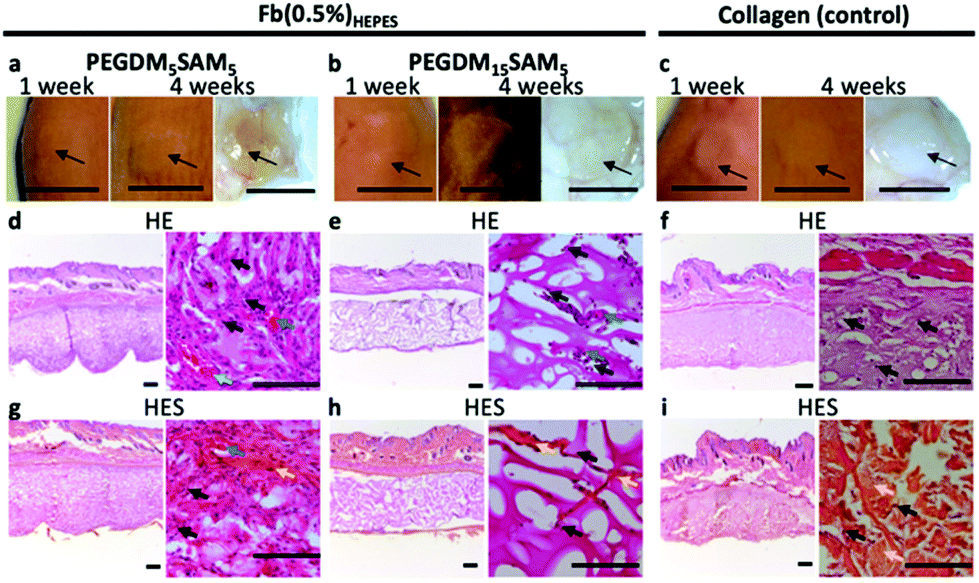 | ||
| Fig. 8 Histological analysis of subcutaneously implanted PEGDM-co-SAM sequential IPN hydrogels. Explanted (a) PEGDM5SAM5 and (b) PEGDM15SAM5 sequential IPN hydrogels made with Fb(0.5%)HEPES following 1 week (left image) and 4 weeks (center image) implantation in mice. (c) Commercially available collagen matrices were implanted as controls. Black arrows show the implants. (d–f) H&E and (g–i) H&ES staining of (d and g) PEGDM5SAM5 and (e and h) PEGDM15SAM5 sequential IPN hydrogels made with Fb(0.5%)HEPES 1 month post-subcutaneous implantations. (f and i) Commercially available collagen matrices were implanted as controls. Staining highlights the macroporous polymeric network of IPN hydrogels (interconnected dark purple network), infiltrated cells (indicated with black arrows), blood vessels (indicated with blue arrows) and collagen deposition (indicated with orange arrows). (d–i, left) Histological images show zoomed out and (d–i, right) zoomed in views. Scale bars: (a–c, left and middle) 1 cm, (a–c, right) 5 mm, (d–i, left) 500 μm, (d–i, right) 50 μm. | ||
Statistical analysis
All values were expressed as mean ± standard deviation (SD). Statistical significance for the mechanical measurements and swelling ratios were determined using a one-way ANOVA test. The different fibrin/PEGDM-co-SAM IPN formulations were compared to cryotreated PEGDM-co-SAM hydrogels individually and to each other. Statistical significance for the metabolic activity measurements were determined using Kruskal–Wallis and Dunn's tests. The different IPN formulations were compared to each other at various time points (1, 3 and 7 days). Differences were considered significant at *p < 0.05, **p < 0.01, and ***p < 0.001.Results and discussion
Synthesis of fibrin/PEGDM-co-SAM IPN hydrogels
In this work, we aimed to engineer new types of macroporous fibrin/PEGDM-co-SAM sequential IPN hydrogels for dermal tissue engineering. IPNs are multicomponent polymeric alloys that combine the unique characteristics of each individual component in an advanced new system.33 Herein, we combined the intrinsic biological properties of fibrin with the excellent mechanical properties of a PEG in macroporous sequential IPN networks.44,45 To this end, PEG and SA were first chemically modified to contain methacrylate residues, allowing formation of radically polymerizable PEGDM and SAM, respectively. The degree of methacrylation for each polymer was quantified by 1H NMR and TNBS assays and was found to be approximately 100% for PEGDM and 67% for SAM (Fig. S1 and S2†). Next, PEGDM and SAM were copolymerized at various concentrations via a free radical polymerization process (Fig. 1). PEGDM-co-SAM hydrogels were subsequently subjected to three consecutive freeze–thaw treatments to introduce interconnected macroporous networks. This process (i.e., cryotreatment), as well as similar techniques such as cryogelation, have been extensively investigated to generate porous biomaterials.44–55 The resulting cryotreated constructs were then rehydrated with various solutions containing fibrin's precursors (fibrinogen and thrombin) and then allowed to polymerize spontaneously. Three different fibrin's precursor solutions were prepared: one solution containing HEPES buffer, the resulting network made with 0.5% w/v fibrin was defined as Fb(0.5%)HEPES, and two other solutions containing deionized water, defined as Fb(0.5%)H2O and Fb(1%)H2O. This sequential fabrication process resulted in the formation of macroporous fibrin/PEGDM-co-SAM IPN hydrogels as shown in Fig. 1.Physical characterization of fibrin/PEGDM-co-SAM IPN hydrogels
The fibrous nature of fibrin networks synthesized in water was most likely a result of the precipitation of fibrin in a non-buffered aqueous solution. These data are particularly promising since the dermis is known to have a fibrous cellular microenvironment.60 The repartition of the two entangled networks, rhodamine-labelled PEGDM-co-SAM (in red) and FITC-labelled fibrin (in green), was then imaged by confocal imaging (Fig. 2c and d). Independently of its concentration, a thin fibrous network of fibrin could be observed through the pores when formulated in water (Fig. 2c and d).
Importantly, regardless of the aqueous solvent or polymer composition used, the various fibrin/PEGDM-co-SAM IPNs fabricated retained the advantageous macroporous structure of the initial cryotreated PEGDM-co-SAM hydrogels. Indeed, sequential polymerization of fibrin did not significantly impact the overall pore size of IPNs which is an essential aspect of tissue engineering scaffolds. This is particularly important for dermal tissue repair, since this process requires sufficient space for cellular colonization and blood vessel formation.56–59,61
For PEGDM15SAM5 hydrogels, QM was approximately 8 for fibrin-free cryotreated PEGDM15SAM5vs. 9.7, 8.4 and 8.7 for fibrin/PEGDM15SAM5 IPN hydrogels when fibrin was formulated in HEPES buffer (0.5% w/v) or in water (0.5% and 1% w/v), respectively (Fig. 3c). The highest swelling ratio observed for IPNs with fibrin made in HEPES buffer could be attributed to a more homogeneous polymerization of fibrin leading to a uniform network when synthesized in a buffered solution, enabling the constructs to absorb and contain more water. It is worth noting that QM for fibrin-free cryotreated PEGDM15SAM5 hydrogels was lower than those fabricated with PEGDM5SAM5 (Fig. 3b and c) showing that increasing the PEG concentration resulted in a decrease in gel swelling. Nevertheless, although the swelling ratio of hydrogels is mainly dictated by the polymer concentration (i.e., crosslinking density), IPN hydrogels tested showed an ability to uptake and retain large amounts of water across the different formulations tested.60 Overall, the swelling ratios measured for the different IPN hydrogels were in the range of those reported in literature, particularly for gels formulated with equivalent concentrations of SA62 or PEG.26
Overall, across all the formulations tested, fibrin hydrogels exhibited weak mechanical properties as previously reported by others,26,28,38 highlighting the importance of engineering fibrin-based hydrogels that are mechanically more stable. We then evaluated the rheological properties of cryotreated PEGDM-co-SAM hydrogels, pre- and post-sequential fibrin fabrication (Fig. 4b and c). Our results confirmed that polymerizing fibrin within cryotreated PEGDM-co-SAM hydrogels significantly increased their elastic moduli (Fig. 4b and c). The G′ was 5.9 kPa for cryotreated PEGDM15SAM5vs. 10.9, 7.8 and 8.2 kPa for fibrin/PEGDM15SAM5 IPNs made with Fb(0.5%)HEPES, Fb(0.5%)H2O and Fb(1%)H2O, respectively (Fig. 4b). Similarly, the G′ was 2.7 kPa for cryotreated PEGDM5SAM5vs. 6.5, 3.7 and 4.8 kPa for fibrin/PEGDM5SAM5 IPNs made with Fb(0.5%)HEPES, Fb(0.5%)H2O and Fb(1%)H2O, respectively (Fig. 4c). Similarly to previous reports,28,30,35,37 the mechanical performance of fibrin/PEGDM-co-SAM IPN hydrogels was enhanced, most likely due to a contribution of both fibrin made in HEPES buffer and the excellent mechanical properties of the PEGDM-co-SAM network itself. Furthermore, as expected and independently of the fibrin formulation tested, the mechanical performance of the PEGDM15SAM5-based hydrogels outperformed those made with PEGDM5SAM5 (Fig. 4b and c). It is also worth noting that, although cryotreatment may compromise the mechanical integrity of polymeric scaffolds,42 cryotreated PEGDM-co-SAM hydrogels retained sufficient mechanical strength to markedly reinforce the fibrin networks for both PEG concentrations tested.
Overall, our data suggested that fibrin-containing IPN hydrogels exhibited much better mechanical integrity (G′ > 3 kPa)26,28,38 than fibrin hydrogels alone (around 0.1 kPa).28,38 The elastic moduli of the macroporous fibrin/PEGDM-co-SAM sequential IPNs were in the range of 0.43–6.6 kPa, which were similar to the mechanical strengths reported for native dermis tissue.68 Furthermore, the superior mechanical characteristics of our engineered IPN hydrogels surpass what others have previously described. For instance, several studies have already reported fibrin-based IPNs combining a fibrin network intertwined with another naturally-derived polymer network such as alginate36 or hyaluronic acid.30,32 However, their elastic moduli did not exceed 0.6 kPa.32 Additionally, other biosynthetic fibrin-based IPNs combining a fibrin network intertwined with a synthetic polymer such as polyvinyl alcohol (PVA)15,16,28,38 or PEG15,16,26 network have also been reported. These fibrin-based materials made via photopolymerization were described to be biocompatible, non-retractable and self-supported.26,28,38 However, their mesoporous network was not compatible with 3D cell culture.15,16,38 Furthermore, UV-exposure during the photopolymerization process could be harmful to mammalian cells.
Biological characterization of fibrin/PEGDM-co-SAM IPN hydrogels
The biomaterials were tested both in vitro and in vivo to assess their biocompatibility, cellular infiltration, and biodegradation.Next, we explored the morphology of HDFs when cultured in the constructs (Fig. 6 and S6†). For each formulation, most fibroblasts adopted their typical spindle-like shape, adhered to the fibrin network and infiltrated the scaffold through their interconnected macropores (Fig. 6 and S6†). These results suggest that the intrinsic properties of fibrin were retained across all the formulations tested and that the macroporous structures of the different hydrogels were favorable for dermal fibroblast culture. Lastly, we investigated whether the scaffolds would promote deposition of collagen I and fibronectin (Fig. 7), two main dermis extracellular matrix (ECM) proteins involved in cell adhesion and protein regulation during wound healing.69,70 Within one week of culture, HDFs were able to secrete and deposit these two proteins across all the biomaterials investigated with no significant difference between them.
Collectively, this set of data suggests that our IPNs form suitable environments for cell infiltration and remodeling.
Additionally, even though SA was incorporated as an enzymatically degradable entity, the biomaterials seemed to remain intact up to 1 month following implantation, suggesting slow biodegradation. This could be attributed to the presence of poly(methyl methacrylate) segments within the polymer network, which are known to poorly degradable in vivo.15,75 Various strategies could be investigated to make these IPN constructs more prone to biodegradation.76 One strategy could be to use biodegradable PEG-based block copolymers (e.g., PEG-co-polyglycolic acid) which contain hydrolysable ester bonds.43,77 Lastly, signs of tissue remodeling were observed with host fibroblasts, de novo synthesis of collagen and early stages of angiogenesis (i.e., neovascularization) within the explanted constructs, indicating good biointegration.14 Therefore, these constructs have great potential for dermal tissue regeneration and wound healing applications.4,5,41,78 However, follow-up validation studies with relevant wound healing models are recommended.
Conclusions
In this study, we report on an innovative synthetic approach to engineer macroporous and interconnected fibrin-based sequential IPN hydrogels for dermal tissue engineering. The engineered IPN hydrogels combined both the superior mechanical properties of PEG and the intrinsic biological features of fibrin. In addition, properties of IPNs can be fine-tuned based on the precursor concentration and solution used during the fabrication process. Their unique macroporous network and biological properties enhanced dermal fibroblast adhesion, infiltration, and proliferation in vitro. Additionally, these IPN hydrogels are biocompatible as demonstrated by a minimal host inflammatory response when implanted subcutaneously in mice. Furthermore, they promoted local tissue remodeling as indicated with host cellular infiltration, de novo ECM synthesis, and neovascularization. Altogether, our data suggest that these macroporous fibrin-based sequential IPN hydrogels have great potential for dermal tissue engineering and other relevant biomedical applications.Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this article.Acknowledgements
This work was supported by the ANR TECSAN Fibriderm (project number ANR-13-TECS-0014) and the Picardy region. Sidi A. Bencherif acknowledges financial support from an NSF CAREER award (DMR 1847843). We wish to thank Fréderic Nadaud for his assistance with the SEM and ESEM characterizations. We also thank Elisabeth Van Hecke for her help with the rheology measurements, Elise Prost for her technical assistance with the NMR measurements, and Ulysse Peirera for his helpful discussions.Notes and references
- R. V. Shevchenko, M. Eeman, B. Rowshanravan, I. U. Allan, I. N. Savina, M. Illsley, M. Salmon, S. L. James, S. V. Mikhalovsky and S. E. James, Acta Biomater., 2014, 10, 3156–3166 CrossRef CAS.
- H. Debels, M. Hamdi, K. Abberton and W. Morrison, Plast. Reconstr. Surg. Glob. Open, 2015, 3, e284 CrossRef.
- L. Ma, C. Gao, Z. Mao, J. Zhou, J. Shen, X. Hu and C. Han, Biomaterials, 2003, 24, 4833–4841 CrossRef CAS.
- A. Francesko, P. Petkova and T. Tzanov, Curr. Med. Chem., 2018, 25, 5782–5797 CrossRef CAS.
- H. Sorg, D. J. Tilkorn, S. Hager, J. Hauser and U. Mirastschijski, Eur. Surg. Res., 2017, 58, 81–94 CrossRef.
- S. P. Zhong, Y. Z. Zhang and C. T. Lim, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 510–525 CAS.
- K. W. Ng, H. L. Khor and D. W. Hutmacher, Biomaterials, 2004, 25, 2807–2818 CrossRef CAS.
- J. Rnjak, Z. Li, P. K. M. Maitz, S. G. Wise and A. S. Weiss, Biomaterials, 2009, 30, 6469–6477 CrossRef CAS.
- J. Rnjak-Kovacina, S. G. Wise, Z. Li, P. K. M. Maitz, C. J. Young, Y. Wang and A. S. Weiss, Biomaterials, 2011, 32, 6729–6736 CrossRef CAS.
- C. Gao, D. Wang and J. Shen, Polym. Adv. Technol., 2003, 14, 373–379 CrossRef CAS.
- B. K. Boekema, M. Vlig, L. O. Damink, E. Middelkoop, L. Eummelen, A. V. Bühren and M. M. W. Ulrich, J. Mater. Sci. Mater. Med., 2014, 25, 423–433 CrossRef CAS.
- Z. Zhu, Y.-M. Wang, J. Yang and X.-S. Luo, Plast. Aesthetic Res., 2017, 4, 219–227 CrossRef.
- H.-M. Wang, Y.-T. Chou, Z.-H. Wen, Z.-R. Wang, C.-H. Chen and M.-L. Ho, PLoS One, 2013, 8, e56330 CrossRef CAS.
- O. Gsib, C. Egles and S. A. Bencherif, J. Mol. Biol. Biotechnol., 2017, 2, 1 Search PubMed.
- O. Gsib, J.-L. Duval, M. Goczkowski, M. Deneufchatel, O. Fichet, V. Larreta-Garde, S. A. Bencherif and C. Egles, Nanomaterials, 2017, 7, 436 CrossRef.
- O. Gsib, M. Deneufchatel, M. Goczkowski, M. Trouillas, M. Resche-Guigon, S. Bencherif, O. Fichet, J.-J. Lataillade, V. Larreta-Garde and C. Egles, IRBM, 2018, 39, 103–108 CrossRef.
- T. V. Anilkumar, J. Muhamed, A. Jose, A. Jyothi, P. V. Mohanan and L. K. Krishnan, Biologicals, 2011, 39, 81–88 CrossRef CAS.
- W. D. Spotnitz, World J. Surg., 2010, 34, 632–634 CrossRef.
- P. A. Janmey, J. P. Winer and J. W. Weisel, J. R. Soc., Interface, 2009, 6, 1–10 CrossRef CAS.
- A. L. Mazlyzam, B. S. Aminuddin, N. H. Fuzina, M. M. Norhayati, O. Fauziah, M. R. Isa, L. Saim and B. H. I. Ruszymah, Burns, 2007, 33, 355–363 CrossRef CAS.
- A. Meana, J. Iglesias, M. Del Rio, F. Larcher, B. Madrigal, M. F. Fresno, C. Martin, F. San Roman and F. Tevar, Burns, 1998, 24, 621–630 CrossRef CAS.
- T. Sun, J. Haycock and S. Macneil, Biomaterials, 2006, 27, 3459–3465 CrossRef CAS.
- Z. J. Rogers, M. P. Zeevi, R. Koppes and S. A. Bencherif, Bioelectricity, 2020, 2, 279–292 CrossRef.
- N. Nakagawa, M. Matsumoto and S. Sakai, Skin Res. Technol., 2010, 16, 137–141 CrossRef.
- P. Matricardi, C. Di Meo, T. Coviello, W. E. Hennink and F. Alhaique, Adv. Drug Delivery Rev., 2013, 65, 1172–1187 CrossRef CAS.
- E. Akpalo, L. Bidault, M. Boissière, C. Vancaeyzeele, O. Fichet and V. Larreta-Garde, Acta Biomater., 2011, 7, 2418–2427 CrossRef CAS.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS.
- L. Bidault, M. Deneufchatel, C. Vancaeyzeele, O. Fichet and V. Larreta-Garde, Biomacromolecules, 2013, 14, 3870–3879 CrossRef CAS.
- O. M. Benavides, J. P. Quinn, S. Pok, J. Petsche Connell, R. Ruano and J. G. Jacot, Tissue Eng., Part A, 2015, 21, 1185–1194 CrossRef CAS.
- F. Lee and M. Kurisawa, Acta Biomater., 2013, 9, 5143–5152 CrossRef CAS.
- P. Losi, E. Briganti, A. Magera, D. Spiller, C. Ristori, B. Battolla, M. Balderi, S. Kull, A. Balbarini, R. Di Stefano and G. Soldani, Biomaterials, 2010, 31, 5336–5344 CrossRef CAS.
- Y. Zhang, P. Heher, J. Hilborn, H. Redl and D. A. Ossipov, Acta Biomater., 2016, 38, 23–32 CrossRef CAS.
- E. S. Dragan, Chem. Eng. J., 2014, 243, 572–590 CrossRef CAS.
- D. Myung, D. Waters, M. Wiseman, P.-E. Duhamel, J. Noolandi, C. N. Ta and C. W. Frank, Polym. Adv. Technol., 2008, 19, 647–657 CrossRef CAS.
- A. Lohani, G. Singh, S. S. Bhattacharya and A. Verma, J. Drug Delivery, 2014, 2014, 583612 Search PubMed.
- A. Shikanov, M. Xu, T. K. Woodruff and L. D. Shea, Biomaterials, 2009, 30, 5476–5485 CrossRef CAS.
- S. L. Rowe and J. P. Stegemann, Biomacromolecules, 2006, 7, 2942–2948 CrossRef CAS.
- L. Bidault, M. Deneufchatel, M. Hindié, C. Vancaeyzeele, O. Fichet and V. Larreta-Garde, Polymer, 2015, 62, 19–27 CrossRef CAS.
- G. Chen, T. Ushida and T. Tateishi, Macromol. Biosci., 2002, 2, 67–77 CrossRef CAS.
- T. M. Freyman, I. V. Yannas and L. J. Gibson, Prog. Mater. Sci., 2001, 46, 273–282 CrossRef CAS.
- A. Amirsadeghi, A. Jafari, L. J. Eggermont, S.-S. Hashemi, S. A. Bencherif and M. Khorram, Biomater. Sci., 2020, 8, 4073–4094 RSC.
- N. Annabi, J. W. Nichol, X. Zhong, C. Ji, S. Koshy, A. Khademhosseini and F. Dehghani, Tissue Eng., Part B, 2010, 16, 371–383 CrossRef CAS.
- S. A. Bencherif, T. M. Braschler and P. Renaud, J. Periodontal Implant Sci., 2013, 43, 251–261 CrossRef CAS.
- S. A. Bencherif, J. A. Sheehan, J. O. Hollinger, L. M. Walker, K. Matyjaszewski and N. R. Washburn, J. Biomed. Mater. Res., Part A, 2009, 90, 142–153 CrossRef.
- S. Lin-Gibson, S. Bencherif, J. M. Antonucci, R. L. Jones and F. Horkay, Macromol. Symp., 2005, 227, 243–254 CrossRef CAS.
- P. Villard, M. Rezaeeyazdi, T. Colombani, K. Joshi-Navare, D. Rana, A. Memic and S. A. Bencherif, Adv. Healthcare Mater., 2019, 8, 1900679 CrossRef.
- A. Memic, M. Rezaeeyazdi, P. Villard, Z. J. Rogers, T. Abudula, T. Colombani and S. A. Bencherif, Macromol. Mater. Eng., 2020, 305, 1900824 CrossRef CAS.
- Z. J. Rogers and S. A. Bencherif, Gels, 2019, 5, 46 CrossRef.
- L. J. Eggermont, Z. J. Rogers, T. Colombani, A. Memic and S. A. Bencherif, Trends Biotechnol., 2020, 38, 418–431 CrossRef CAS.
- A. Memic, T. Colombani, L. J. Eggermont, M. Rezaeeyazdi, J. Steingold, Z. J. Rogers, K. J. Navare, H. S. Mohammed and S. A. Bencherif, Adv. Ther., 2019, 2, 1800114 CrossRef.
- K. Joshi-Navare, L. J. Eggermont, Z. J. Rogers, H. S. Mohammed, T. Colombani and S. A. Bencherif, in Racing for the Surface: Pathogenesis of Implant Infection and Advanced Antimicrobial Strategies, ed. B. Li, T. F. Moriarty, T. Webster and M. Xing, Springer International Publishing, Cham, 2020, pp. 511–542 Search PubMed.
- J. Kim, S. A. Bencherif, W. A. Li and D. J. Mooney, Macromol. Rapid Commun., 2014, 35, 1578–1586 CrossRef CAS.
- S. Kennedy, S. Bencherif, D. Norton, L. Weinstock, M. Mehta and D. Mooney, Adv. Healthcare Mater., 2014, 3, 500–507 CrossRef CAS.
- M.-E. Han, S.-H. Kim, H. D. Kim, H.-G. Yim, S. A. Bencherif, T.-I. Kim and N. S. Hwang, Int. J. Biol. Macromol., 2016, 93, 1410–1419 CrossRef CAS.
- M. Rezaeeyazdi, T. Colombani, A. Memic and S. A. Bencherif, Materials, 2018, 11, 1374 CrossRef.
- B. Dhandayuthapani, Y. Yoshida, T. Maekawa and D. S. Kumar, Int. J. Polym. Sci., 2011, 2011, 290602 Search PubMed.
- V. Sharma, N. Patel, N. Kohli, N. Ravindran, L. Hook, C. Mason and E. García-Gareta, Biomed. Mater., 2016, 11, 055001 CrossRef.
- C. Faure and G. Barry, Monit. Hosp., 2011, 240, 33–44 Search PubMed.
- C. S. L. Müller, C. Schiekofer, R. Körner, C. Pföhler and T. Vogt, J. Dtsch. Dermatol. Ges., 2013, 11, 537–548 Search PubMed.
- O. Okay, in Hydrogel Sensors and Actuators, Springer, Berlin, Heidelberg, 2009, pp. 1–14 Search PubMed.
- S. MacNeil, Mater. Today, 2008, 11, 26–35 CrossRef CAS.
- V. Abbate, X. Kong and S. S. Bansal, Enzyme Microb. Technol., 2012, 50, 130–136 CrossRef CAS.
- J. M. Zuidema, C. J. Rivet, R. J. Gilbert and F. A. Morrison, J. Biomed. Mater. Res., Part B, 2014, 102, 1063–1073 CrossRef.
- N. A. Kurniawan, T. H. S. van Kempen, S. Sonneveld, T. T. Rosalina, B. E. Vos, K. A. Jansen, G. W. M. Peters, F. N. van de Vosse and G. H. Koenderink, Langmuir, 2017, 33, 6342–6352 CrossRef CAS.
- H. E. Davis, S. L. Miller, E. M. Case and J. K. Leach, Acta Biomater., 2011, 7, 691–699 CrossRef CAS.
- N. Laurens, P. Koolwijk and M. P. M. De Maat, J. Thromb. Haemostasis, 2006, 4, 932–939 CrossRef CAS.
- H. Phillips , Nat. News, 1998.
- B. Holt, A. Tripathi and J. Morgan, J. Biomech., 2008, 41, 2689–2695 CrossRef.
- K. Wolf, S. Alexander, V. Schacht, L. M. Coussens, U. H. von Andrian, J. van Rheenen, E. Deryugina and P. Friedl, Semin. Cell Dev. Biol., 2009, 20, 931–941 CrossRef CAS.
- S. Stenman and A. Vaheri, J. Exp. Med., 1978, 147, 1054–1064 CrossRef CAS.
- R. Shah, M. C. Pierce and F. H. Silver, J. Biomed. Mater. Res., Part A, 2017, 105, 15–22 CrossRef CAS.
- D. Wolfram, B. Oberreiter, C. Mayerl, E. Soelder, H. Ulmer, H. Piza-Katzer, G. Wick and A. Backovic, Immunol. Lett., 2008, 118, 96–100 CrossRef CAS.
- D.-J. Li, K. Ohsaki, K. Ii, P.-C. Cui, Q. Ye, K. Baba, Q.-C. Wang, S. Tenshin and T. Takano-Yamamoto, J. Biomed. Mater. Res., 1999, 45, 322–326 CrossRef CAS.
- D. Bakker, C. A. van Blitterswijk, S. C. Hesseling, J. J. Grote and W. T. Daems, Biomaterials, 1988, 9, 14–23 CrossRef CAS.
- R. G. Hill, in Biomaterials, Artificial Organs and Tissue Engineering, ed. L. L. Hench and J. R. Jones, Woodhead Publishing, 2005, pp. 37–47 Search PubMed.
- S. Lin-Gibson, S. Bencherif, J. A. Cooper, S. J. Wetzel, J. M. Antonucci, B. M. Vogel, F. Horkay and N. R. Washburn, Biomacromolecules, 2004, 5, 1280–1287 CrossRef CAS.
- S. A. Bencherif, A. Srinivasan, J. A. Sheehan, L. M. Walker, C. Gayathri, R. Gil, J. O. Hollinger, K. Matyjaszewski and N. R. Washburn, Acta Biomater., 2009, 5, 1872–1883 CrossRef CAS.
- A. Grada, J. Mervis and V. Falanga, J. Invest. Dermatol., 2018, 138, 2095–2105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01161d |
| This journal is © The Royal Society of Chemistry 2020 |