
Induction of a higher-ordered architecture in glatiramer acetate improves its biological efficiency in an animal model of multiple sclerosis†
 ‡ab
Yee Ming
Khaw,
‡ac
Lazaro A.
Pacheco,
b
Zhengzhong
Tan,
b
Kaimin
Cai,
b
Jianjun
Cheng
*b
and
Makoto
Inoue
*ac
‡ab
Yee Ming
Khaw,
‡ac
Lazaro A.
Pacheco,
b
Zhengzhong
Tan,
b
Kaimin
Cai,
b
Jianjun
Cheng
*b
and
Makoto
Inoue
*ac
Abstract
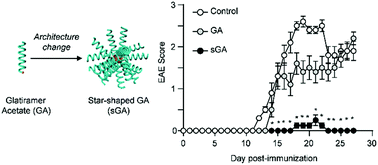
Glatiramer acetate (GA), a linear random copolypeptide, is a first-line treatment for multiple sclerosis (MS). A major concern, however, is that GA treatment is associated with adverse effects and poor patient adherence due to the need for frequent injections. Here we describe improved performance of the polymeric drug, even at low doses with less-frequent injections, through the modification of its architecture into a star-shaped GA (sGA). In a sGA, multiple GAs are covalently linked onto a core, which greatly changes their properties such as molecular weight, size, and shape. The spherical sGA is retained longer in the body after intraperitoneal injection, and is more readily internalized by RAW 264.7 macrophage cells and bone marrow-derived dendritic cells than GA. In C57BL/6 mice induced with experimental autoimmune encephalitis, a mouse model for MS, sGA treatment exerts disease amelioration effect that is significantly better than that of GA despite a lower dose and less frequent injection. Moreover, spinal cord pathologies of demyelination and leukocyte infiltration are dramatically less pronounced in the sGA treatment condition compared to the GA treatment condition. Thus, we propose that sGA with a higher-ordered architecture offers an attractive and potentially viable treatment option for MS patients.
 Please wait while we load your content...
Please wait while we load your content...

