
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of degradable magnesium on paracrine signaling between human umbilical cord perivascular cells and peripheral blood mononuclear cells†
Qian
Wang
 ,
Lei
Xu
,
Heike
Helmholz
,
Regine
Willumeit-Römer
and
Bérengère J. C.
Luthringer-Feyerabend
*
,
Lei
Xu
,
Heike
Helmholz
,
Regine
Willumeit-Römer
and
Bérengère J. C.
Luthringer-Feyerabend
*
Institute of Materials Research, Division for Metallic Biomaterials, Helmholtz-Zentrum Geesthacht (HZG), Geesthacht, Germany. E-mail: Berengere.Luthringer@hzg.de
First published on 25th September 2020
Abstract
Human mesenchymal stem cells (MSC) interact with numerous immune cells that can promote regenerative processes and inhibit inflammatory responses. We hypothesised that the cross-talk between human umbilical cord perivascular cells (HUCPV; an alternative source of MSC) and peripheral blood mononuclear cells (PBMC) could be influenced by degradable transwell magnesium (Mg). To study the correlations between paracrine signaling and specific cellular behaviour during the host response to Mg, we used a transwell coculture system for up to 7 days. The proliferation and viability of both cell types were not significantly influenced by Mg. When HUCPV were cultured with degradable Mg, a moderate inflammation (e.g., lower secretions of pro-inflammatory interleukin 1 beta and IL2, and tumour necrosis factor alpha, interferon gamma, anti-inflammatory interleukins 4, 5, 10, 13, and 1 receptor antagonists and granulocyte colony stimulating factor), and an increased pro-healing M2 macrophage phenotype were observed. Moreover, when PBMC were cultured with degradable Mg, the expression of migration/wound healing related cytokines (interleukin 8, granulocyte-macrophage colony-stimulating factor, monocyte chemoattractant protein 1 and macrophage inflammatory protein 1α/β) was upregulated, accompanied by an increase in the migration ability of HUCPV (cell scratch assay). In addition, an increased pro-osteogenic potential was demonstrated via an increase of osteoblastic markers (e.g., alkaline phosphatase activity, specific gene expression and cytokine release). These results collectively imply that Mg possesses osteo-immunomodulatory properties. They also help to design Mg-based bone substitute biomaterials capable of exhibiting desired immune reactions and good clinical performance.
1. Introduction
Magnesium (Mg) and its alloys have been investigated as degradable implants for cardiovascular,1,2 orthopaedic3–5 and bone regenerative applications6,7 because of their desirable bioactive properties, such as stimulated osteoblast adherence8 and formation of cartilage.9 Usually, immune responses to a biomaterial comprise non-specific inflammation.10 Peripheral blood mononuclear cells (PBMC), comprising lymphocytes (T cells, B cells, and natural killer cells; 70–90%), monocytes (10–20%) and dendritic cells (1–2%), have been widely used as immune cells to evaluate the in vitro inflammatory reaction to biomaterials.11,12 In addition, PBMC are able to secrete a broad range of cytokines in non-specific inflammatory responses, including pro-inflammatory interleukins 1 beta (IL1β), IL2, tumour necrosis factor alpha (TNFα), and interferon gamma (IFNγ),13 as well as anti-inflammatory cytokines, such as IL10.14 As an in vitro model to follow the initial immune reaction after biomaterial implantation, the secretion of cytokines by PBMC could be very informative. The interactions between the immune system and biomaterials rely on the tissue surrounding the implant, which will induce the tissue-specific innate immune response and the subsequent adaptive immune response.15 After biomaterial implantation, when the blood–biomaterial come into contact, immediately a layer of proteins from the surrounding microenvironment could be adsorbed onto the material surface, forming a surface matrix and attracting specific cells from the innate immune system (natural killer cells, dendritic cells, mast cells, and granulocytes (neutrophils, basophils and eosinophils), and macrophages) to the damaged sites in short time. These immune cells secrete cytokines and chemokines that are capable of recruiting tissue repair cells, such as mesenchymal stem cells (MSC) etc., into the injured sites. Normally, the occurring immune response is accompanied by tissue repair reactions in the following 2–10 days.16 Although a variety of cell types are involved in tissue repair, macrophages, a small group of PBMC, play a key role in the initial immune response by secreting cytokines and chemokines that directly affect tissue repair.17 As a source of MSC, the human umbilical cord perivascular cells (HUCPV) were selected. HUCPV exhibit a low immunogenicity18,19 and potent immunomodulatory effects,20 and a natural primary potential to differentiate into an osteogenic phenotype.21–23 The cell-to-cell communication between immune cells and MSC could regulate the immune reaction via paracrine signaling, in which a cell could secrete the factors to influence the behaviour and functions of nearby cells.24 Therefore, in the current study, the molecule releases were measured after 1, 4, and 7 days to follow the initial immune reaction to the degradable Mg material.Promoting tissue regeneration by modulating the immune system using biomaterials is one of the strategies followed in regenerative medicine and in smart biomaterial development. Thus, as immune responses cannot be avoided and are necessary in the early stages of bone implantation, a moderate inflammation and an increase of the pro-healing M2 macrophage phenotype around the implants may be more efficient to promote fracture healing and lasting stability of bone implants.25–28 In recent years, numerous reports have proposed that Mg and its alloys play a role in the modulation of immune systems.29,30 For instance, in vitro studies have demonstrated the direct role of Mg in decreasing the pro-inflammatory cytokine production,31 as well as mediating the anti-inflammatory effects.32 The anti-inflammatory effects of Mg salts33 or nanoparticles34 were confirmed in vivo. To date, research studies have confirmed that a faster inflammation resolution stimulated by Mg and Mg-based materials could be desirable to convert the macrophages from the M0 to the pro-healing M2 phenotype in vitro.35,36 In addition, it has become clear that mesenchymal stem cells (MSC) also possess immunomodulatory properties that can modulate several immune cells,37 especially macrophages.38,39 For instance, coculture of MSC was able to induce macrophages to adopt an enhanced regulatory (M2) phenotype via increased levels of IL4, IL10, and IL13, and reduced concentrations of TNFα, IFNγ and IL2.40 Increasing numbers of clinical tests show that the success of MSC-based immunomodulation relies on the assessment of molecule secretion and their interaction with immune cells.41 Also, MSC can influence the various pathways of immune responses in a paracrine manner.42,43 Furthermore, a recent study about Mg ions (magnesium sulphate) has indicated that Mg could modulate some immunoregulatory properties of MSC.44 However little is reported about the role of degradable Mg in the immunomodulatory properties of MSC in a biomaterial scenario. In particular, the effects of degradable Mg on paracrine signaling between MSC and immune cells require further examination.
Indeed, the communication of MSC–immune cells is not a one-way street. Immune cells have the potential to attract stem cells to the damaged tissue and several mediators have been implicated in the migration of MSC. Among these factors, monocyte chemoattractant protein 1 (MCP-1 also known as C–C motif chemokine ligand 2 or CCL2), macrophage inflammatory protein (MIP-1α/CCL3), IL-8 and TNFα45,46 can be listed. In addition, macrophage polarisation and release of cytokines and other substances influence tissue healing and remodelling.47 These macrophages and their polarised morphological variants always stay at the material–tissue interface in vivo. This cell–cell fusion also contributes to the degradation of biomaterials via their phagocytosis and released mediators.48,49 The released mediators also play crucial roles in MSC differentiation. For instance, pro-inflammatory cytokines (TNFα and IL1β) inhibited the osteogenic differentiation of stem cells.50 Soluble factors, e.g., granulocyte-macrophage colony-stimulating factor (GM-CSF), enhanced osteogenic differentiation in Saos-2 cells51 or stem cells.52 The secretions of basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), and IL12 could also be markers of osteoblastic differentiation.23 In addition, numerous reports over recent years have demonstrated that Mg ions can stimulate osteogenic differentiation of mesenchymal stem cells (MSC) and bone regeneration in vitro53,54 and Mg-based alloys can promote bone formation in vivo.6,55 Thus, the influence of degradable Mg on immune cell-mediated MSC behaviour (e.g., migration/wound healing and osteogenic differentiation), notably the paracrine signaling, needs further examination.
To answer the above questions (influences of degradable Mg on paracrine signaling between immune cells and MSC) in an interactive microenvironment, a two-way communication, transwell (TW) system, was used.56 By employing the TW system, HUCPV and PBMC (and especially their derived macrophages; later referred to as PBMC/macrophages in this manuscript) were grown separately while sharing the same microenvironment and soluble factors. Thus, the contribution of each cell type can be investigated. Initially, the current study focused on the role of Mg degradation in paracrine secretions in relation to inflammation, as well as the M2 macrophage phenotype. Besides, the secreted cytokines, which are responsible for MSC behaviour (recruitment and osteogenic differentiation), were determined. Several involved aspects, wound healing assay and pro-osteogenic activity of HUCPV, were also examined.
2. Experimental
2.1. Cell isolation and culture
HUCPV were derived from the perivascular site (Wharton's jelly) of umbilical cords. Umbilical cord samples were obtained from Asklepios Klinik Altona (Hamburg, Germany) immediately after caesarean sections of consenting donors. The HUCPV isolations were approved by the Ethics Committee of the Hamburg Medical Association (PV4058) and were performed as previously described.57 The purity of the isolated HUCPV was validated via immunophenotyping by flow cytometry (S3e; Bio-Rad Laboratories, Munich, Germany) presented in the ESI (Fig. S1a†). The immunophenotype of HUCPV was characterised using CD105 (SH2) and CD90 (Thy-1) as mesenchymal cell positive and specific markers,22 and CD5458 and CD3159 to exclude the contamination of fibroblast and endothelial cells, respectively. Additionally, the HUCPV morphology (Fig. S1b†) was evaluated via live/dead staining with calcein AM (living cells, green) and ethidium homodimer-1 (dead cells, red; 1 and 2 μM, respectively; Thermo Fisher Scientific, Schwerte, Germany) using an inverted fluorescence microscope (Eclipse Ti, Nikon GmbH, Düsseldorf, Germany). The isolated HUCPV were expanded in an α-minimum essential medium (α-MEM; Invitrogen, Fisher Scientific GmbH, Schwerte, Germany) containing 15% (v/v) foetal bovine serum for human mesenchymal stem cells (FBS; Biological Industries, Beit-Haemek, Israel) and 1% (v/v) penicillin/streptomycin (P/S; Life Sciences, Karlsruhe, Germany) under physiological conditions (5% CO2, 20% O2, 95% relative humidity, and 37 °C). The medium was changed every 2–3 days and the cells were cultured until approximately 80% confluence was reached. HUCPV at 5–8 passages were used for the biological tests.Human leukocyte-enriched blood samples were provided by the University Hospital Hamburg-Eppendorf (UKE; Hamburg, Germany), and then PBMC were isolated using the Ficoll Paque 400 (GE Healthcare, Chicago, Illinois, USA) from the buffy coat as described by the former protocol.60 Meanwhile, the human plasma (above the buffy coat) was collected, centrifuged at 300g for 10 min, passed through a 0.45 μm sterile filter (Thermo Fisher Scientific, Schwerte, Germany) and frozen at −20 °C for further use. The isolated PBMC subpopulations used in this study are shown in Fig. S2 (ESI†).
Here we used pooled allogeneic human plasma instead of FBS consistent with other research studies.61,62 Because of the species difference of biological response modifiers (BRM) between humans and bovines, using autologous human plasma is a more appropriate way to test the effects of biomaterials on human PBMC/macrophages and HUCPV.63 Additionally, compared to human plasma, FBS might contain higher background of non-specific cell stimulatory factors.64,65 Importantly, to avoid a very high level of variability between human serum or plasma lots,66 we used pooled human plasma throughout our project. Therefore, the α-MEM medium was supplemented with 10% human plasma (v/v) and 1% P/S and used as the cell culture medium in our system.
2.2. Material preparation
Biomaterials were produced at Helmholtz-Zentrum Geesthacht, Geesthacht, Germany. High-purity Mg (99.93%) was prepared by permanent mould gravity casting, followed by a T4 treatment of the produced ingots, and extrusion into rods of 10 mm diameter. Finally, after being machined to obtain a diameter of 9 mm, Mg discs were cut with 1.5 mm thickness and each disc weight was about 0.2 g. After cleaning via sonication for 20 min in 100% n-hexane, 100% acetone and 100% ethanol as well as sterilisation in 70% ethanol (Merck KGaA, Darmstadt, Germany), each disc was preincubated for 24 hours in 500 μL of cell culture medium (the same as above) prior to use.2.3. TW systems
In order to elucidate how the interaction between PBMC/macrophages and HUCPV is influenced by Mg biomaterial in vitro, a transwell (5 μm pore size; Alvetex™ Strata, Glasgow, United Kingdom) was used (Fig. 1a). An agarose coating prevents cell adhesion and survival on a tissue culture plate. In an agarose-coated 12-well plate (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany), HUCPV were seeded at a density of 2 × 105 cells (25 μL) on preincubated Mg discs for 30 min (lower compartment or well bottom), and then 1.8 mL of fresh cell culture medium was added. Afterwards, PBMC were seeded at a density of 5 × 105 cells in 0.2 mL of culture medium in the upper compartment. Single culture of HUCPV and PBMC and coculture without Mg disc were selected as controls. To detect the possible artefact or background caused by the cell culture medium and Mg disc, the pure cell culture medium and supernatant from Mg discs without cells were regarded as blank and no-cell controls, respectively (all experimental conditions and their abbreviations are presented in Table 1). In the context of transplantation, biomaterials and the surrounding cells, such as cells of innate immune response (e.g. monocytes and macrophages) and MSC, interplay with each other during the first two weeks.16 Considering the validation of the present TW system (data not shown), all systems were cultured up to 7 days. The medium was renewed on day 4. The supernatants and cells were collected on day 1, 4 and 7 and stored at −80 °C until use.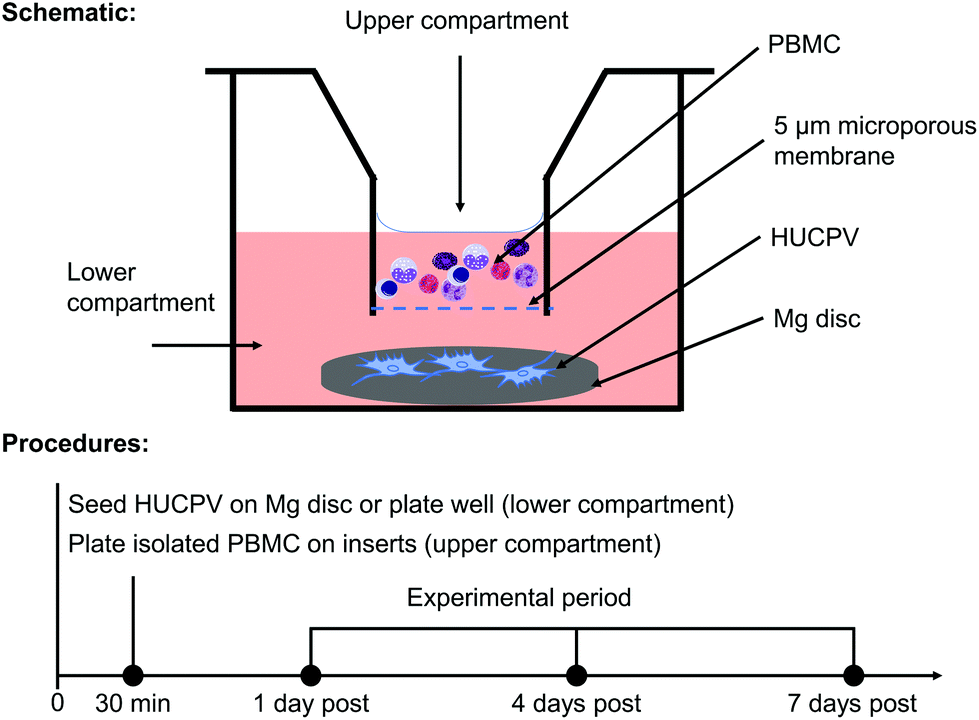 | ||
| Fig. 1 In vitro TW coculture system. | ||
| No. | Abbreviation | Experimental condition | |
|---|---|---|---|
| TW system | 1 | Blank | Cell culture medium |
| 2 | Mg | Only Mg disc | |
| 3 | H | HUCPV alone | |
| 4 | H + Mg | HUCPV on Mg disc | |
| 5 | P | PBMC alone | |
| 6 | P + Mg | PBMC (upper compartment) with Mg disc (lower compartment) | |
| 7 | H + P | Coculture of HUCPV and PBMC | |
| 8 | H + P + Mg | Coculture of HUCPV on Mg disc (lower compartment) with PBMC (upper compartment) |
2.4. pH and Mg concentration
The pH values and Mg contents under two different experimental conditions were quantified at each time point using an ArgusX pH Meter (Sentron Europe BV, Roden, Netherlands) and by atomic absorption spectroscopy (AAS; Agilent 240/280 Series AA (Agilent Technologies, Waldbronn, Germany)), respectively.2.5. Cell viability and proliferation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 (v/v) in DNA dilution buffer (2.5 M NaCl in 19 mM sodium citrate, pH 7). The diluted sample of volume 100 μL was transferred into the wells of a 96-well-plate (flat bottom, black microtiter plate) and measured in duplicate. Then 50 μL of DNA working buffer (2 M NaCl in 15 mM sodium citrate, pH 7) and 50 μL of bisbenzimide solution (2 μg mL−1) were added and the plates were incubated for 15 min in the dark. The fluorescence emission (excitation wavelength, 355 nm and emission wavelength, 460 nm) was measured with a VICTOR3 multilabel microtiter plate reader (PerkinElmer, Massachusetts, USA). The DNA content was calculated using the fluorescence values of a corresponding standard curve. All chemicals were purchased from Sigma-Aldrich Chemie GmbH (Taufkirchen, Germany).
:5 (v/v) in DNA dilution buffer (2.5 M NaCl in 19 mM sodium citrate, pH 7). The diluted sample of volume 100 μL was transferred into the wells of a 96-well-plate (flat bottom, black microtiter plate) and measured in duplicate. Then 50 μL of DNA working buffer (2 M NaCl in 15 mM sodium citrate, pH 7) and 50 μL of bisbenzimide solution (2 μg mL−1) were added and the plates were incubated for 15 min in the dark. The fluorescence emission (excitation wavelength, 355 nm and emission wavelength, 460 nm) was measured with a VICTOR3 multilabel microtiter plate reader (PerkinElmer, Massachusetts, USA). The DNA content was calculated using the fluorescence values of a corresponding standard curve. All chemicals were purchased from Sigma-Aldrich Chemie GmbH (Taufkirchen, Germany).
2.6. Production of cytokines
To identify the potential secretory production involved in the crosstalk between HUCPV and PBMC/macrophages, the protein concentrations of IL1β, IL2, IL4, IL5, IL8, IL10, IL12, IL13, IL1RA, TNFα, IFNγ, granulocyte colony stimulating factor (G-CSF), GM-CSF, MCP-1, MIP-1α/β, bFGF, and VEGF were measured in the supernatants using a Bio-Plex multiplex immunoassay (M500KCAF0Y; Bio-Rad Laboratories, Munich, Germany) according to the manufacturer's instructions. 50 μL of bead solution was added to the wells of a 96-well plate, then mixed (Eppendorf thermomixer comfort, Hamburg, Germany) with 50 μL of diluted standards, blanks and samples and finally incubated at room temperature (RT) with shaking at 850 rpm for 30 min. Afterwards, 25 μL of detection antibody was added and incubated for 30 min at RT with shaking at 850 rpm. Then 50 μL of streptavidin-phycoerythrin (PE) was added and incubated for 10 min at RT with shaking at 850 rpm. The assays were ended and resuspended in 125 μL of assay buffer and shaken at 850 rpm for 30 s. In each shaking step, the plate was covered with a new sheet of stealing tape and protected from light. All plates were read using a Bio-Plex 200 system (Bio-Rad Laboratories, Munich, Germany) and analysed using Bio-Plex Manager (Bio-Rad Laboratories, Munich, Germany; version 6.1). The variation induced by cell numbers under each condition was normalised by the DNA content.2.7. Flow cytometry
To address the variation in the monocyte/macrophage ratios in single culture or coculture with and without Mg disc, the M2 phenotype was investigated by flow cytometry. The harvested cells were washed twice with ice cold PBS. After 1000 rpm centrifugation for 5 min, the cells were resuspended at a density of 1 × 106 cells in 1 mL of PBS supplemented with 1% bovine serum albumin (BSA; Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany). The antibody anti-human CD 163-PE (scavenger receptor cysteine-rich type 1 protein M130, a high affinity receptor for the hemoglobin–haptoglobin complex; 560933, BD Biosciences, Heidelberg, Germany) was diluted 10 times in PBS. The cells were incubated with the diluted antibody for 1 h on ice with gentle shaking in the dark. Then the cells were washed and fixed with 3.7% formaldehyde (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) and permeabilised with 0.7% Tween 20 (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany). The cells were washed with PBS and stained with the intracellular antibody anti-human CD 68-fluorescein isothiocyanate (FITC; scavenger receptor class D, member 1; 130096964, Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) for 30 min at RT with shaking in the dark. The cells were washed again and resuspended with 1% BSA in PBS. Anti-human CD 68+ was selected as the pan marker for macrophages67 and anti-human CD 163+68 as the surface marker for M2. The expressions of CD 68 and CD 163 were investigated using a flow cytometer. The flow cytometry panel was designed as follows. Positive groups were cells stained with anti CD 68 and anti CD 163 to determine the subpopulation of type 2 macrophages in CD 68 positive cells. Single staining of CD 68 and CD 163 was performed for further result compensation. Propidium iodide (PI) drop solution (Bio-Rad Laboratories, Munich, Germany) was used to exclude the dead cells after each analysis. Isotype controls were the corresponding mouse IgG2b (130099119, Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) and IgG1 (559320, Thermo Fisher Scientific, Schwerte, Germany). Cells resuspended in 1% BSA in PBS were used as the blank control to determine the forward scatter (FSC) and side scatter (SSC) parameters during the assays. The proportion of M2-macrophages within the monocyte population (“M2/CD 68+”) was calculated as follows: (CD 68+ CD 163+)/CD 68+ cells (%).
2.8. Wound healing assay
To clarify whether the migratory ability of HUCPV was affected by the Mg levels in the supernatants and/or stimulatory cytokines from PBMC/macrophages, wound healing or scratch assay of HUCPV was carried out under different conditions. HUCPV were seeded at a density of 1 × 105 cells per well in 12-well plates containing α-MEM supplemented with 15% FBS until the cell monolayer was formed. Then the cells were starved in an α-MEM medium with low serum (1% FBS). After 24 h, the cells were treated with a 0.01 mg mL−1 mitomycin C (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) solution to prevent further cell proliferation.69–71 The scratch was created with a 1 mL pipette tip. The cells were treated for another 24 h with cell culture medium (α-MEM containing 10% plasma) as the control. The TW (day 1) and CM supernatants collected under Mg, P, P + Mg conditions were also used. The scratch gap was documented using an inverted microscope (Eclipse Ti, Nikon GmbH, Düsseldorf, Germany) at t0 and t24 h under bright light conditions. The relative cell migration distance was calculated from the difference of the widths of the scratch gaps between 0 and 24 h using NIS-Elements imaging software (Eclipse Ti, Nikon GmbH, Düsseldorf, Germany).2.9. Pro-osteogenic potential of HUCPV
| Full name | Abbreviation | GenBank no. | Sequences | Annealing (°C) |
|---|---|---|---|---|
| Collagen type I alpha 1 | COL1A1 | NM_000088 | Forward: 5′-AAGACATCCCACCAATCACC-3′ | 60 |
| Reverse: 5′-GCAGTTCTTGGTCTCGTCAC-3′ | ||||
| Osteocalcin | OC | NM_199173 | Forward: 5′-ATGAGAGCCCTCACACTCCT-3′ | |
| Reverse: 5′-TGGACACAAAGGCTGCAC-3′ | ||||
| Osteopontin | OPN | NM_000582 | Forward: 5′-CTCCATTGACTCGAACGACTC-3′ | |
| Reverse: 5′-CAGGTCTGCGAAACTTCTTAGAT-3′ | ||||
| B2 microglobulin | B2M | NM_004048.2 | Forward: 5′-TGCTGTCTCCATGTTTGATGTATCT-3′ | |
| Reverse: 5′-TCTCTGCTCCCCACCTCTAAGT-3′ | ||||
| Glyceraldehyde3-phosphate dehydrogenase | GAPDH | NM_002046.7 | Forwards: 5′-GTCGGAGTCAACGGATTTG-3′ | |
| Reverse: 5′-TGGGTGGAATCATATTGGAA-3′ | ||||
| Ribosomal protein L10 | RPL10 | NM_006013.4 | Forwards: 5′-AGTGGATGAGTTTCCGCTTT-3′ | |
| Reverse: 5′-ATATGGAAGCCATCTTTGCC-3′ |
2.10. Statistical analysis
Overall, at least two wells per condition with technical duplicate of three different donors of HUCPV and PBMC were performed (n = 6). The results in the section “pH and Mg concentration” were obtained from three individual experiments with two replicates (n = 3). The results were presented as arithmetic mean ± standard deviation (SD). The significances between the two groups were investigated by the t-test. The statistical significances between multiple comparisons were analysed by one-way ANOVA or one-way ANOVA on ranks using the SigmaPlot (Systat software GmbH, Erkrath, Germany; version 13.0). The following post hoc multiple comparisons were based on Tukey's when equal variance was assumed or Dunn's when equal variance was not assumed (α = 0.05, *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001). For RT-qPCR, to detect the gene expression, the Volcano Plot was used in which the regulation threshold and P value were set as 1.5 and 0.05, respectively (calculated using CFX Manager Software version 3.1; Bio Rad, Munich, Germany).3. Results
3.1. pH and Mg concentration
To characterise the experimental conditions, the pH values and Mg contents of the supernatants at each time point were analysed (Table 3). According to the pH values under all experimental conditions, Mg and/or cells were exposed to pH ranging from 8.05 to 8.32. The Mg contents under conditions without Mg disc were in a similar range (0.60 to 0.78 mM) to the value (0.71 mM) under blank conditions. Considering the treatment with the Mg disc, the Mg contents under cell-seeded conditions (i.e. H + Mg, P + Mg and H + P + Mg) fell in the ranges from 4.71 to 6.87, 15.62 to 18.03, and 8.19 to 14.86 mM at various time points, respectively.| Conditions | pH | Mg contents (mM) | ||||
|---|---|---|---|---|---|---|
| Day 1 | Day 4 | Day 7 | Day 1 | Day 4 | Day 7 | |
| Mg | 8.27 ± 0.004 | 8.20 ± 0.013 | 8.30 ± 0.054 | 7.66 ± 0.074 | 11.75 ± 0.079 | 8.36 ± 0.044 |
| H | 8.09 ± 0.012 | 8.05 ± 0.029 | 8.09 ± 0.013 | 0.68 ± 0.007 | 0.63 ± 0.005 | 0.60 ± 0.002 |
| H + Mg | 8.15 ± 0.041 | 8.27 ± 0.030 | 8.24 ± 0.009 | 5.74 ± 0.001 | 18.03 ± 0.070 | 13.81 ± 0.206 |
| P | 8.05 ± 0.009 | 8.09 ± 0.023 | 8.14 ± 0.015 | 0.54 ± 0.002 | 0.64 ± 0.004 | 0.78 ± 0.004 |
| P + Mg | 8.23 ± 0.022 | 8.31 ± 0.066 | 8.23 ± 0.036 | 4.71 ± 0.011 | 15.62 ± 0.064 | 8.19 ± 0.057 |
| H + P | 8.05 ± 0.020 | 8.09 ± 0.014 | 8.25 ± 0.132 | 0.64 ± 0.001 | 0.70 ± 0.003 | 0.69 ± 0.003 |
| H + P + Mg | 8.27 ± 0.051 | 8.32 ± 0.039 | 8.26 ± 0.058 | 6.87 ± 0.043 | 17.91 ± 0.083 | 14.86 ± 0.107 |
3.2. Cell viability and proliferation
Cell live/dead staining was performed to estimate the cell viability and adherence/proliferation behaviour. Bright green represented the living cells, whereas red fluorescence indicated the dead cells (Fig. 2a and c). The nucleic acid content can be a quantifiable indicator of the cell number.72 The bisbenzimide can specifically bind to the nucleic acid. Therefore, the fluorescence emission of their complexes correlates with the cell number and reflect the cell proliferation.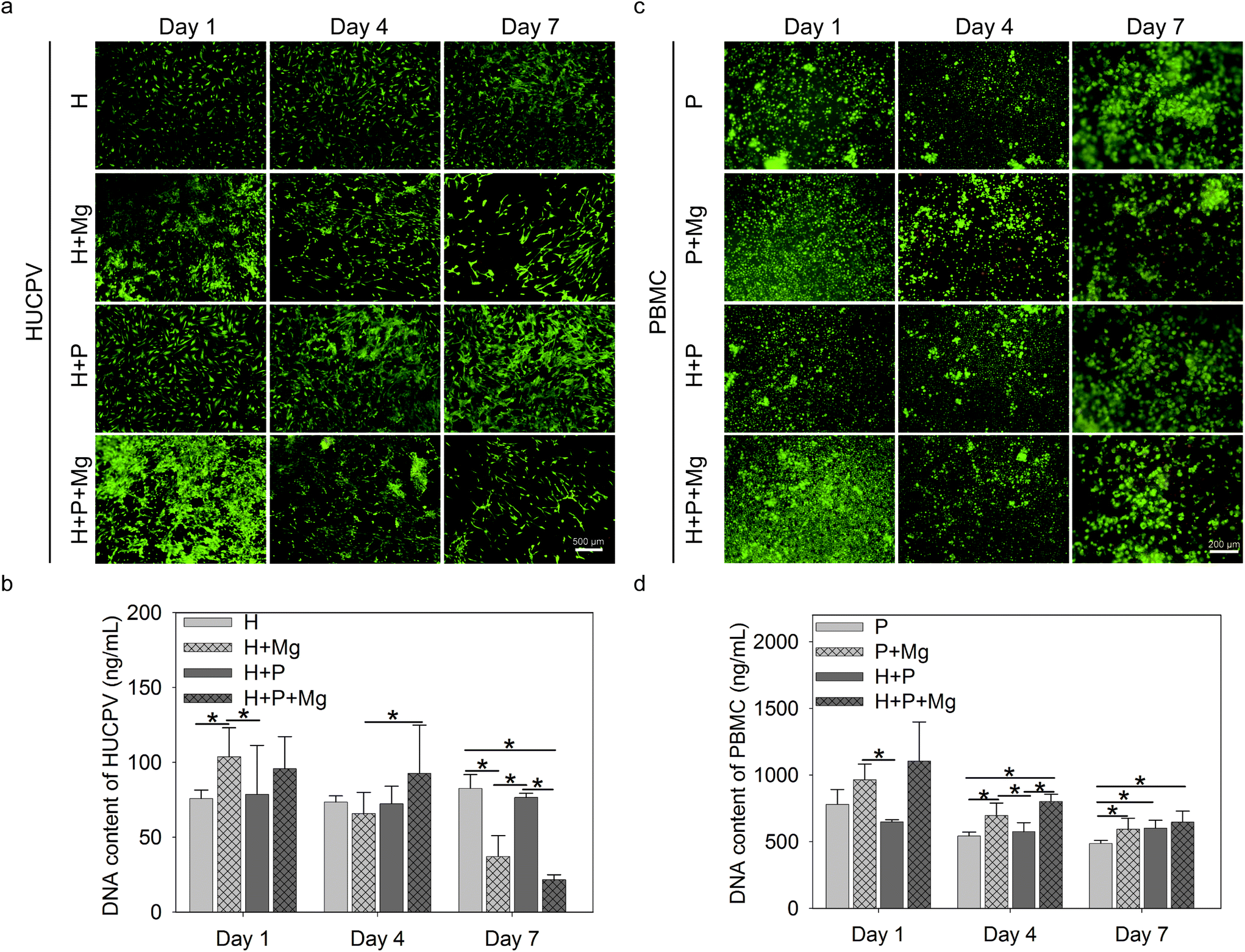 | ||
| Fig. 2 Cell viability and proliferation of HUCPV and PBMC in the TW system. (a) Live/dead staining of HUCPV and (c) PBMC. Green colour and red spot represent alive and dead cells, respectively. (b) DNA content of HUCPV and (d) PBMC. Scale bars represent 500 μm (a) and 200 μm (c). Stars indicate significant differences between the two conditions (ANOVA; α = 0.05, *p ≤ 0.05). H: HUCPV; P: PBMC. | ||
Adherent HUCPV could be found under all experimental conditions up to 7 days of culture (Fig. 2a, green). After 1 day of culture, the Mg disc increased the cell number (Fig. 2a) and DNA contents (Fig. 2b) of HUCPV compared to the HUCPV monolayer. In the subsequent culture period, PBMC stimulated the HUCPV growth under H + Mg vs. H + P + Mg (day 4) and H + Mg vs. H + P (day 7) conditions, rather than Mg discs. Particularly, a reduction was observed on day 7 when HUCPV were directly exposed to the Mg surfaces. Besides, HUCPV and PBMC mixed cultures, HUCPV seeded on the Mg disc with and without PBMC or kept as monocultures, showed no significantly increased number of dead cells and proliferative cells compared with controls. Again, similar observations were made with the corresponding DNA contents.
Besides, green-labelled adherent PBMC were identified, as shown in Fig. 2c, in the presence of the Mg disc, the dead cells were not apparently increased, and meanwhile, the cell number of PBMC increased on day 4 and 7 in the presence of Mg, H or both(Fig. 2d). Specifically, PBMC exposed to Mg could proliferate much faster than those cultured with HUCPV on day 1 and 4.
3.3. Roles of degradable Mg in the immunomodulatory properties of HUCPV
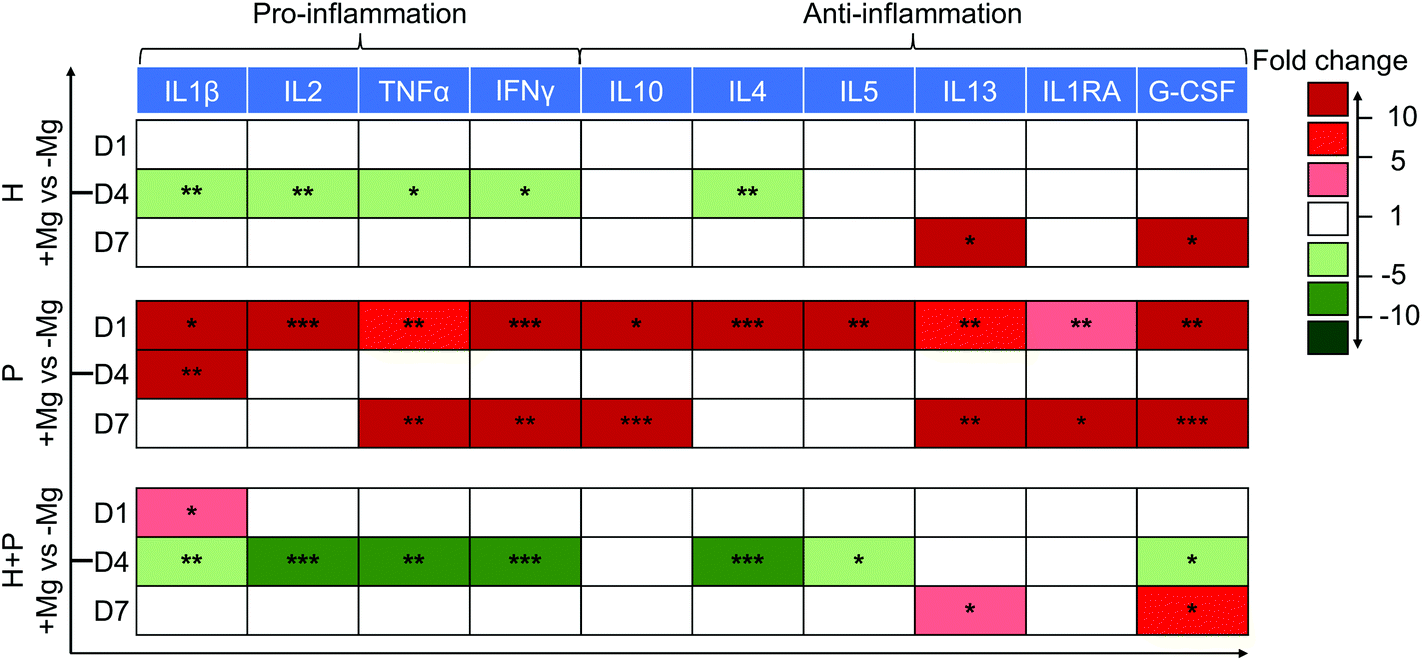 | ||
| Fig. 3 Cytokine levels in the supernatant of HUCPV, PBMC and HUCPV–PBMC coculture in the presence and absence of Mg disc. The colour scale represents fold change compared between the conditions with Mg disc and controls without Mg disc (red: up-regulation; white: no change; and green: down-regulation). Asterisks indicate significant differences between the two conditions (t-test; α = 0.05, *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001). H: HUCPV; P: PBMC. | ||
A decrease in the levels of pro-inflammatory proteins (IL2, TNFα, IL1β and IFNγ) on day 4 and an increase in the production of anti-inflammatory (IL13 and G-CSF) cytokines on day 7 were observed in HUCPV in contact with Mg. Thus, Mg could induce HUCPV to an anti-inflammatory state, especially on day 4.
Initially, PBMC were activated when cultured with Mg (day 1), expressing high levels of all the above-mentioned cytokines. However, when PBMC were cocultured with HUCPV, a downregulation of pro-inflammatory IL2, TNFα, and IFNγ (fold change −5 to −10) on day 4, and an increase in the anti-inflammatory IL13 and G-CSF levels (fold change 5 to 10) on day 7 were observed (Fig. 3). Besides, a similar increase of IL4/5 caused by Mg was also observed in PBMC. However, a clear decrease of IL4/5 caused by Mg was observed in cocultures. Obviously, a moderate inflammation could be observed, when HUCPV mediates Mg-induced cytokine production in PBMC.
Taken together, these results indicated that when exposed to Mg, HUCPV could create an anti-inflammatory microenvironment. Furthermore, HUCPV play a role in the suppression of Mg-induced inflammatory cytokine production in PBMC. The immune-modulation of HUCPV on PBMC/macrophage behaviour is mainly dominated by the immunomodulatory characteristics of HUCPV.73,74
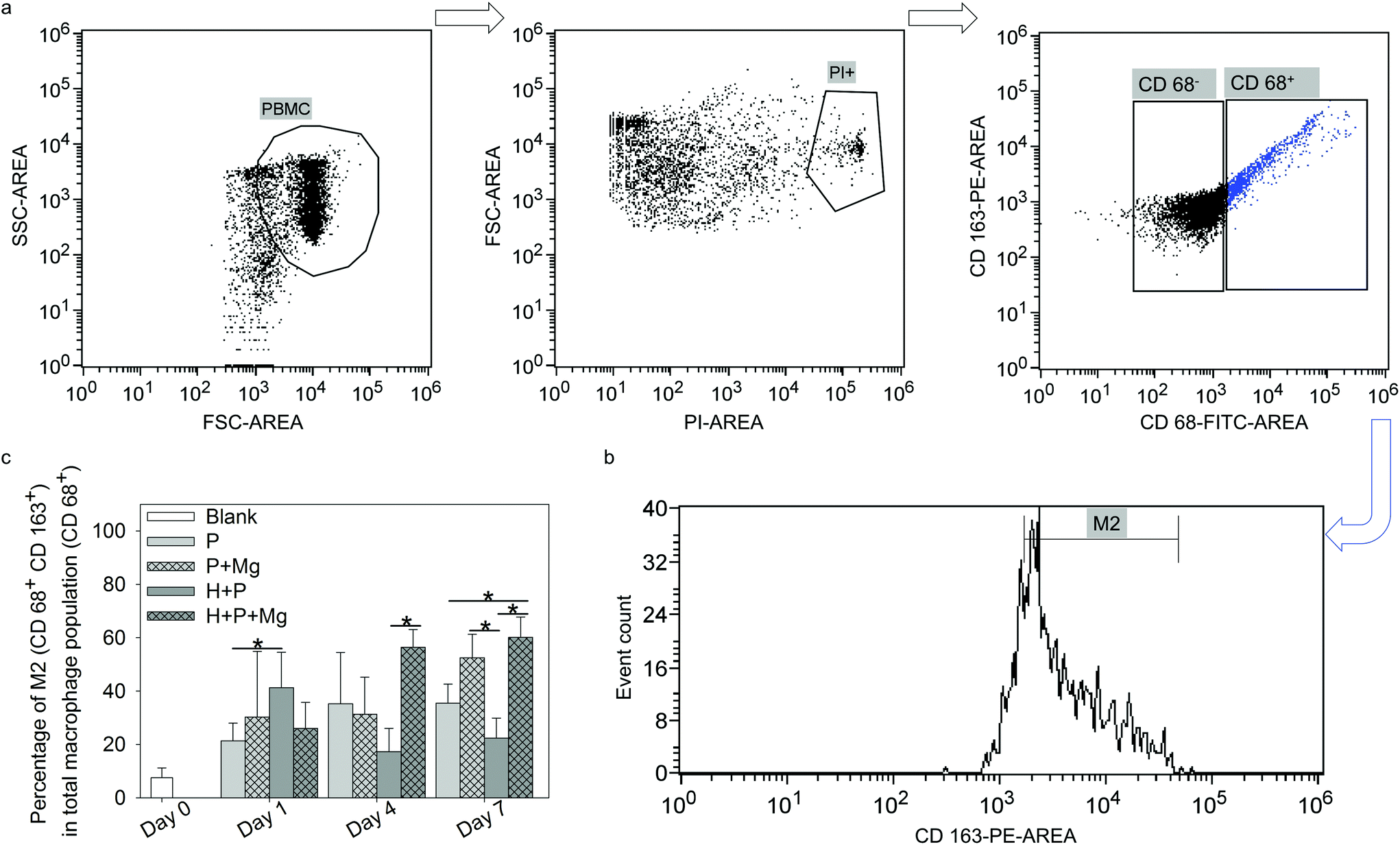 | ||
| Fig. 4 Flow cytometry analysis of the M2 subpopulation at each time point in the TW system. (a) The selection of macrophage (CD 68+) cells. In brief, the living cells were firstly gated out, and then the CD 68+ cells were gated out. (b) The selection of M2 macrophages in the CD 68+ subgroup, in which, CD 163 was used as the specific marker to identify M2 macrophages from total macrophages. (c) Percentage of M2 in the total macrophage population. Stars indicate significant differences between the two conditions (ANOVA; α = 0.05, *p ≤ 0.05). H: HUCPV; P: PBMC. | ||
3.4. Roles of degradable Mg in inflammatory secretion-mediated fracture healing
Within the context of tissue repair, after recruitment of stem cells by immune cells at the site of injury, the cell–cell interactions between immune cells and stem cells are vital. Thus the contributions of immune cells (PBMC) to HUCPV behaviour under the stimulation of Mg were studied, including wound healing assay and the evaluation of HUCPV osteogenic potential.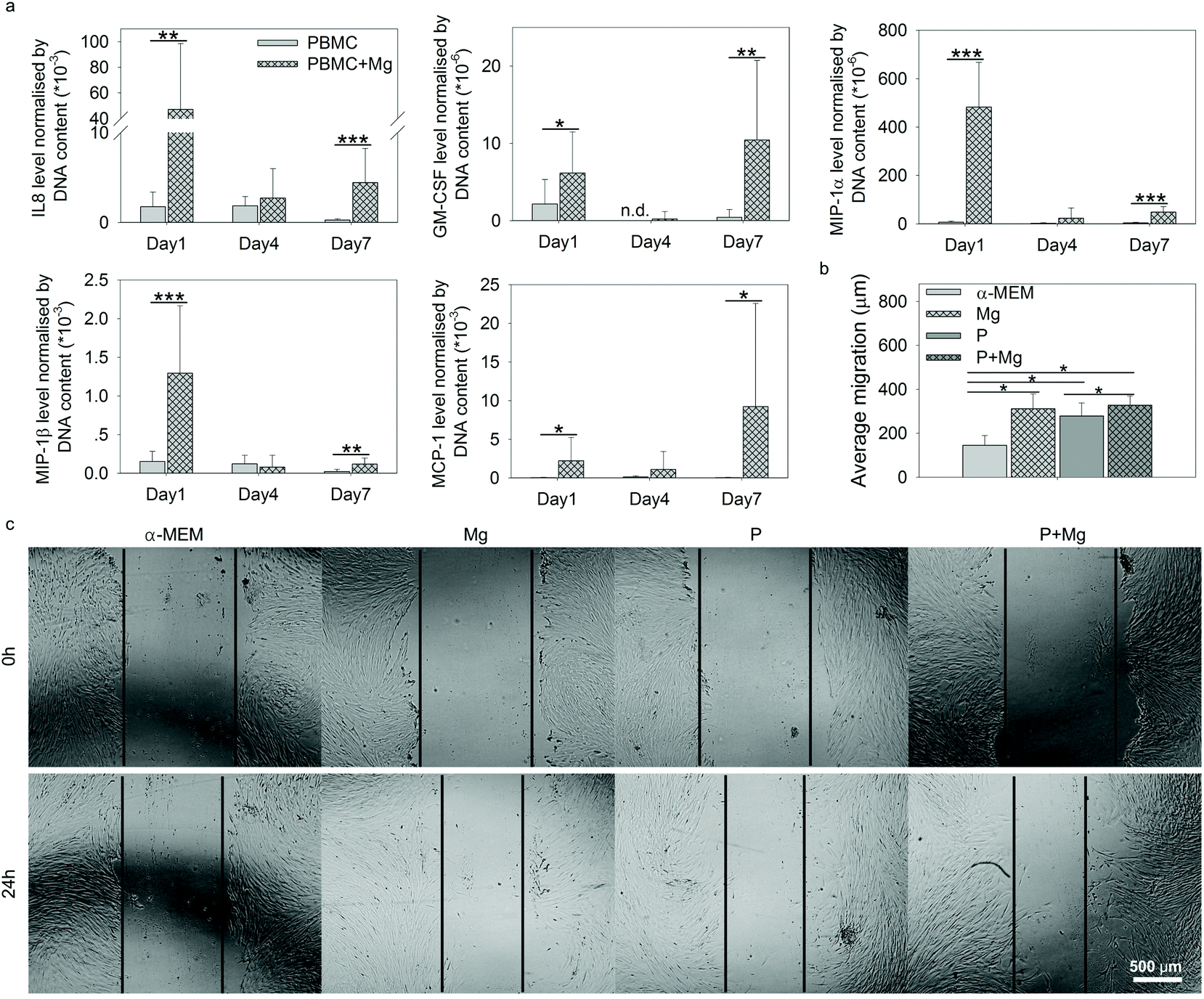 | ||
| Fig. 5 The migratory ability of HUCPV. (a) Selected cytokine (IL8, GM-CSF, MIP-1α/β, and MCP-1) production under P and P + Mg conditions at each time point. (b) Average migration distance of HUCPV, when cultured in the supernatant from the TW system. The migration distance was calculated from the area without cell difference between 0 and 24 h. (c) Microscopic examination of a scratch assay. The scratch at 0 and 24 h were quantified by the gap width. Scale bar represents 500 μm. Stars indicate significant differences between the two conditions (t-test or ANOVA, α = 0.05; *p ≤ 0.05; **p ≤ 0.01; and ***p ≤ 0.001). P: PBMC. | ||
The higher levels of these selected 5 cytokines in PBMC (+Mg) indicated the possible promotion of MSC migration into injured sites,45,75,76 to stimulate the wound healing process. Thus, attempting to further examine the migration potency of HUCPV affected by Mg, an in vitro wound healing assay was carried out with the supernatants collected on day 1 under the TW conditions, comprising Mg, P, and P + Mg.
The migration distance of HUCPV was calculated from the scratch area difference between 0 and 24 h (Fig. 5b). In comparison with the α-MEM control group, Mg or PBMC (P) or both (P + Mg) could significantly increase the migratory ability of HUCPV. Similarly, a stimulated migration capacity of HUCPV was observed under the P + Mg condition, compared with HUCPV under the P condition. A large amount of HUCPV existed at and crossed the edge of scratch in TW (Fig. 5c). These results revealed that HUCPV have more capacity to migrate when cultured with Mg and/or PBMC for 24 h.
As shown in Fig. 6a, Mg elevated the production of bFGF (H vs. H + Mg on day 1), VEGF (H vs. H + Mg or H + P vs. H + P + Mg on day 7), and IL12 (H vs. H + Mg on day 7). Interestingly, on day 1 or day 4, the involvement of PBMC could significantly reduce the production of these three cytokines (H vs. H + P or H + Mg vs. H + P + Mg).
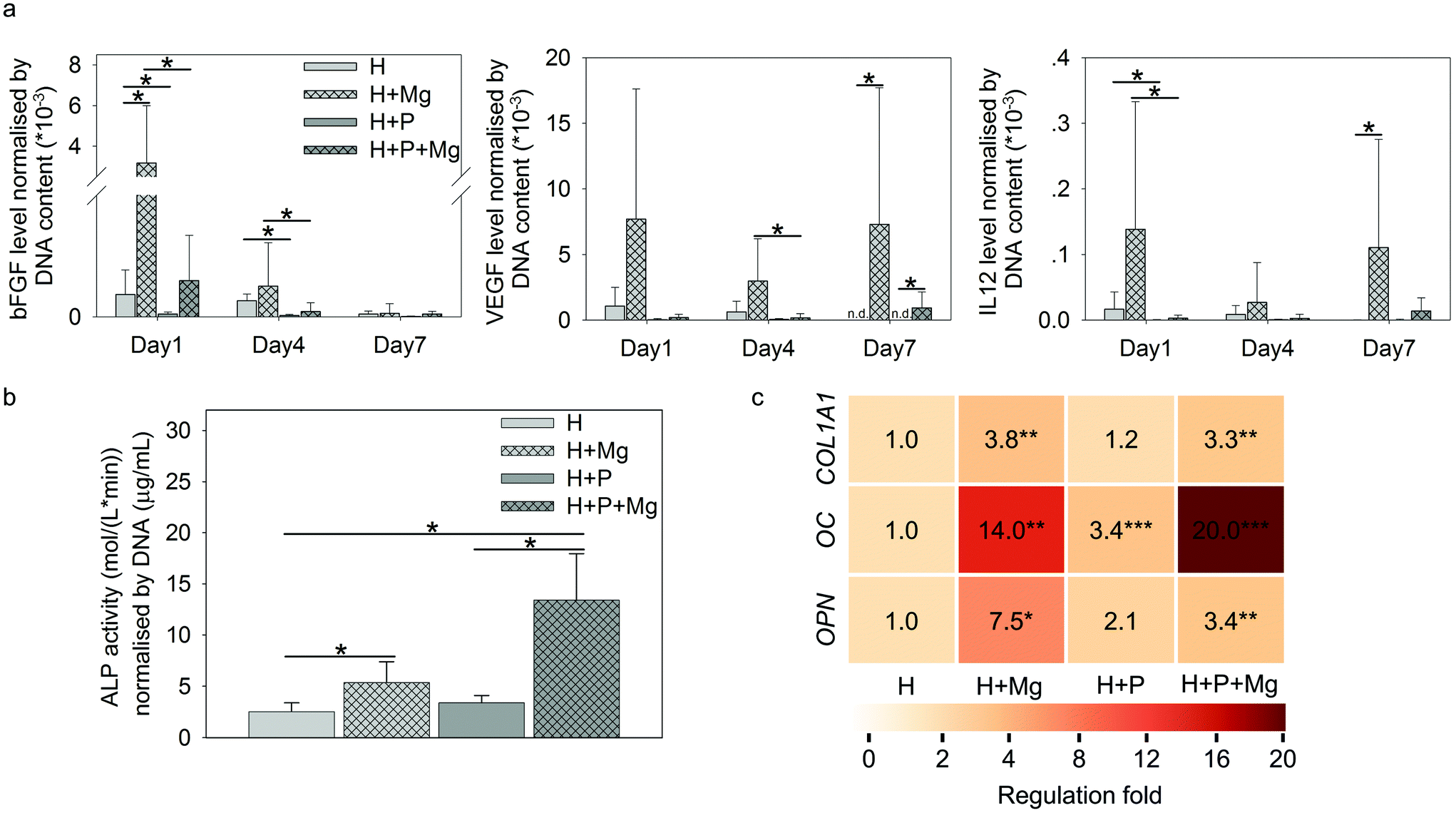 | ||
| Fig. 6 Pro-osteogenic activity of HUCPV. (a) Selected cytokine (BFGF, VEGF, and IL12) production under H/H + P with and without Mg at each time point. (b) ALP activity in TW on day 7. (c) Osteogenic gene expressions in TW on day 7. Cytokine production and ALP activity were normalised by the DNA content. Fold of gene expression was compared to controls in TW. The significance is represented by asterisks and obtained from post hoc multiple comparisons between each condition or comparing to controls (H) (t-test or ANOVA, α = 0.05; *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001). H: HUCPV; P: PBMC. | ||
The ALP activity normalised by DNA is presented in Fig. 6b. The ALP activity on day 7 can be attributed to HUCPV as PBMC/macrophages do not produce a significant amount of ALP.77
In TW, compared to the HUCPV monoculture or cocultures, the levels of ALP were significantly increased when the cells were exposed to the Mg disc. In addition, the fold change of osteogenesis-related gene expression of COL1A1, OC and OPN in HUCPV is indicated by the colour scale in Fig. 6b. The coculture with PBMC could enhance the gene expression of OC in HUCPV. Similarly, an increase of COL1A1, OC and OPN was found when Mg was used. Even a synergistic effect of P and Mg could be seen for OC (the regulation fold reached 20).
4. Discussion
A biomaterial triggers host immune reactions after implantation prior to MSC recruitment.16,78 MSC can interact with immune cells by modulating immune responses and promoting bone regeneration (osteo-immunomodulation), and indeed MSC and immune cells can share the secreted cytokines.41,42 In addition, the immune responses can affect the mechanical properties and performance of biomaterials.79 However, a comprehensive knowledge of the effects of degradable Mg on paracrine signaling between immune cells (PBMC) and MSC is still lacking. Thus, the roles of degradable Mg in the immunomodulatory properties of MSC, as well as immune-mediated MSC behaviour, like MSC migration and osteogenic differentiation, were studied in the current research.Tissue repair cells, immune cells, and Mg ions should be the three protagonists involved in the cascades of degradable Mg-modulated fracture healing. HUCPV are defined as mesenchymal progenitors, multipotent and allogeneic stem cells.22 In comparison with bone marrow mesenchymal stem cells (BMMSC), HUCPV, as an alternative foetal MSC, possess the differentiation capacity and the immunomodulatory properties as well.20–23 Macrophages are one of the key immune cells (PBMC) participating in fracture healing thanks to their polarisation plasticity.38 Mg and Mg-based alloys have been proved to be potential degradable materials for regenerative applications.80–82 Here pure Mg was selected to avoid an increase in the biological complexity due to an alloying element. As a more representative human in vivo-like system of biomaterial-modulated wound healing, TW is highly relevant for a microenvironment and allows an in-depth tracking of the effects of degradable Mg on cell–cell interactions. In the TW system, PBMC were typically seeded on the top of the chamber and HUCPV in the lower compartment, and thus the different cells were always interacting with each other, allowing monocytes to differentiate into macrophages while HUCPV can differentiate into osteoblast-like cells. Meanwhile, the Mg contents varied from 0.5 mM to 18 mM as a consequence of Mg degradation. Thus, in the context of Mg degradation, the focus of this study could provide an insight into paracrine secretions between HUCPV and PBMC during the fracture healing process.
In most mammalian cells, the total concentration of Mg is in the range of 14–20 mM, and that in an intracellular medium is between 0.5 and 0.7 mM.83,84 Moreover, many cells could maintain the Mg contents at the physiological levels, which is irrelevant to the extracellular Mg concentration.85 In the present TW system, the Mg concentration in the cell culture medium (α-MEM) was 0.7 mM. As reported, Mg2+ could play various roles in the cell growth: stimulation of the cell proliferation86 or inhibitory effect on the cell number.87 For instance, in U2OS cells, a slight inhibition of cell proliferation was observed when the Mg concentration was higher than 5 mM.88 Other studies suggested that Mg ions can significantly inhibit the cell growth in the range from 12.5 to 50 mM.89–93 Similarly, as calculated in Fig. 2b, an observation was concluded that there is a decreased DNA content of HUCPV in the presence of Mg (around 14 mM), while an increased DNA content of HUCPV when exposed to Mg and P + Mg (6 mM or about 18 mM), separately. Meanwhile, Mg at various concentrations ranging from 0.54 to 17.91 mM promoted PBMC proliferation (Fig. 2d). Thus, the effects of Mg ions on cellular proliferation could be biphasic94 or rely on the whole microenvironment between materials and cells.
In the host immune response after Mg implantation, the secretory cytokines were the vital players in the regulation of inflammation. Because of the pro- and anti-inflammatory properties of MSC and PBMC/macrophages,40,95 this correlation was evaluated via the cytokine production of the inflammatory panel (TNFα, IFNγ, IL1β, IL2, IL4, IL5, IL10, IL13, IL1RA, and G-CSF) in the current TW coculture. In the presence of Mg, HUCPV secreted a lower number of pro-inflammatory TNFα, IFNγ, IL1β, and IL2 on day 4, when the Mg contents in the supernatants reached 18 mM. Moreover, on day 1 and day 7 (the Mg concentration varied in the range from 5 mM to 8 mM), Mg-stimulated PBMC exhibited an intensive inflammation state, releasing higher levels of pro- and anti-inflammatory cytokines, and this stimulation probably could be recognised by the inflammatory response and cell activation.96 Meanwhile, Mg, at a concentration of 16 mM, had no effects on the inflammatory cytokine production in PBMC (day 4). Therefore, the effects of degradable Mg on the production of inflammatory cytokines in HUCPV monoculture or PBMC was biphasic. Nevertheless, the coculture of HUCPV could switch such Mg-induced higher levels of pro-inflammatory cytokines to a down-expressed state on day 1 (Mg: 7 mM) and day 4 (Mg: 18 mM). Similarly, on day 7, at a concentration of 15 mM, the Mg-induced higher secretions of anti-inflammatory cytokines in PBMC was also attenuated by HUCPV (colour scale: dark red to light red). These comparisons of cocultures indicated the immuno-suppressive role of HUCPV interacting with PBMC under the stimulation of Mg, regardless of the Mg concentration. Thus, the interface of Mg biomaterial–tissue could retain a more moderate status of inflammation. This is consistent with the study of the immuno-suppressive capacity of MSC that MSC can regulate the transition from a T helper type 1 (Th1)-driven response to an anti-inflammatory Th2 response, characterised by lower concentrations of TNFα, IFNγ and IL2, as well as higher production of IL4, IL5, IL10 and IL13.40
Macrophages are polarised by various stimuli to generate heterogeneous populations with different characteristics and functions. The main subtypes of macrophages are M1 and M2 phenotype macrophages, which are vital factors in the resolution of inflammation and tissue remodelling.97,98 M1 phenotype macrophages, which are associated with pro-inflammatory activities,99 are often induced by toll-like receptor (TLR) ligands or Th1 cytokines (TNFα and IFNγ).100 In addition, the M2 phenotype macrophages are polarised by Th2-derived cytokines, including IL4, IL10, IL13, transforming growth factor beta (TGFβ) or prostaglandin E2 (PGE2).100 M2 macrophages are called “tissue repair macrophages” because they promote tissue repair through immune tolerance and tissue remodelling, removal of debris, and immune regulation.101 Besides, MSC present interactive but complicated immunomodulatory properties for immune cells via their cytokines. In the scale of MSC immunomodulation in cells from innate and adaptive immune responses,102,103 despite various cell groups, i.e., monocytes, lymphocytes and dendritic cells, many research studies consolidated that immunomodulation by MSC is crucial for macrophage polarisation.100,101 For instance, MSC was indicated to trigger the monocyte population towards the pro-healing M2 phenotype,104,105 which aids in the resolution of inflammation and tissue remodelling.97,98,106 In addition, the correlation between Mg2+ and MSC and its effects on bone tissue have been investigated.60,107,108 The anti-inflammatory properties of Mg were reported as well.109,110 In the past decades, multiple studies have confirmed that MSC could induce anti-inflammatory M2 polarisation,38,111,112 which could depend on paracrine signaling through the secretion of soluble factors, such as TGFβ, PGE2 and indoleamine 2,3-dioxygenase (IDO).113–115 The current results of TW suggested that the subpopulation of M2 was increased by Mg alone or Mg cocultured with HUCPV (±Mg). It implied that the M2 macrophage phenotype was not only influenced by Mg, but also modulated by MSC (HUCPV). This effect of MSC is at least partially mediated by soluble factors, such as the widely reported PGE2.116,117 Prostaglandin E synthase (PTGES) is a key enzyme in the production of PGE2.118 Our preliminary work has shown a significantly upregulated expression of PTGES2 in HUCPV with magnesium on day 7 (ESI, Fig. S3†). This result implies that the PGE2 production could be increased by Mg even though the HUCPV proliferation was decreased by Mg. The role of Mg in influencing PGE2 expression in immunomodulation was also certified by a study that Mg2+ (5 mM) could regulate immune responses by decreasing the expression of IL1β and IL6, and by increasing the expression of PGE2 and IL10 in Murine C3H/10T1/2 MSC stimulated with LPS or TNFα.44 Furthermore, the secretory ability of MSC is not correlated with their proliferative ablity.119 Therefore, the decreased DNA contents of HUCPV on day 7 (Fig. 2b) should not be directly linked to their M2 phenotype polarisation potential.
During the fracture healing process, MSC initially migrate to the bone injury site to participate, before differentiating into osteoblasts-like cells.120–123 This recruitment of MSC can be influenced by MCP-1, MIP-1α, MIP-1β and IL8.45,124 As shown in Fig. 5a, cytokines inducing MSC migration, such as IL8, GM-CSF, MCP-1, and MIP-1α/β in PBMC, were dramatically upregulated by Mg, especially on either day 1 or day 7. In addition, the supernatants from Mg or PBMC or both could stimulate HUCPV migration into the closure area in TW (Fig. 5b and c). Therefore, the enhanced migratory ability of HUCPV was not only due to Mg itself (Mg contents), but also due to the involvement of Mg-induced paracrine signaling of IL8, GM-CSF, MCP-1, and MIP-1α/β. Furthermore, the increased recruitment of HUCPV could also contribute to the understanding of in vivo increased bone formation observed after magnesium implantation6,125–127 and even the increased osteoblast apposition on the implant surface.8,128 Moreover, the higher levels of IL12, bFGF, and VEGF are recognized as markers of foetal-derived osteoblastogenesis.23 Mg-Stimulated higher secretions of IL12, bFGF, and VEGF, as well as various osteogenic parameters such as ALP activity, collagen synthesis and expression of OC and OPN, indicated that Mg can enhance the osteogenic differentiation of MSC.129,130 As reported, monocytes can enhance the osteogenic gene expression of MSC131 and further promote osteoblast formation as well.132,133 Furthermore, it has also been well-documented that macrophages may engage in the degradation of biomaterials and can contribute to the osteogenic behaviour of MSC which aid in bone regeneration.134 The desired immune microenvironment can effectively stimulate osteogenesis, and therefore, the roles of degradable Mg via paracrine secretions in immune-mediated osteogenesis would need to be further studied for longer periods (as 7 days reflect the early osteogenesis).
5. Conclusions
Our findings emphasised degradable Mg influenced the HUCPV–PBMC interaction. Mg could affect the cell behaviour in cooperation with other cell types. Mg collaborated with HUCPV to attenuate inflammation via a moderate cytokine release, accompanied by the pro-healing M2 macrophage phenotype. In turn, Mg in synergy with immune cells (PBMC) could stimulate HUCPV migration, while the pro-osteogenic potential of HUCPV could be influenced by Mg but not PBMC. Thus, a suppressive inflammation and faster bone tissue repair could be expected due to the osteo-immunomodulatory properties of degradable Mg.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
Qian Wang was funded by the China Scholarship Council (CSC; 201608210187). We appreciate the University Medical Centre Hamburg-Eppendorf, Hamburg, Germany, for blood donation.Written informed consent from the donor was obtained for the use of these samples in research and all experiments were conducted in accordance with the Declaration of Helsinki. HUCPVC were isolated and all experiments were performed with the approval from the local ethical committee Ethik-Kommission der Ärztekammer Hamburg (Hamburg, Germany, PV4058).
Notes and references
- W. Zhou, Y. Zheng, M. Leeflang and J. Zhou, Acta Biomater., 2013, 9, 8488 Search PubMed.
- T. Schilling, M. Bauer, L. LaLonde, H. J. Maier, A. Haverich and T. Hassel, in Magnesium Alloys, ed. M. Aliofkhazraei, InTech, 2017, ch. 7, p. 191 Search PubMed.
- S. Kamrani and C. Fleck, BioMetals, 2019, 32, 185 Search PubMed.
- M. P. Staiger, A. M. Pietak, J. Huadmai and G. Dias, Biomaterials, 2006, 27, 1728 Search PubMed.
- R. Radha and D. Sreekanth, J. Magnesium Alloys, 2017, 5, 286 Search PubMed.
- T. Kraus, S. F. Fischerauer, A. C. Hänzi, P. J. Uggowitzer, J. F. Löffler and A. M. Weinberg, Acta Biomater., 2012, 8, 1230 Search PubMed.
- A. Brown, S. Zaky, H. Ray Jr. and C. Sfeir, Acta Biomater., 2015, 11, 543 Search PubMed.
- H. Zreiqat, C. Howlett, A. Zannettino, P. Evans, G. Schulze-Tanzil, C. Knabe and M. Shakibaei, J. Biomed. Mater. Res., Part A, 2002, 62, 175 Search PubMed.
- M. Shimaya, T. Muneta, S. Ichinose, K. Tsuji and I. Sekiya, Osteoarthr. Cartil., 2010, 18, 1300 Search PubMed.
- B. D. Ratner, A. S. Hoffman, F. J. Schoen and J. E. Lemons, Biomaterials science: an introduction to materials in medicine, Elsevier, San Diego, 2nd edn, 2004 Search PubMed.
- R. Chen, J. Curran, F. Pu, Z. Zhuola, Y. Bayon and J. A. Hunt, Polymers, 2017, 9, 254 Search PubMed.
- A. Hammerl, C. E. Diaz Cano, E. M. De-Juan-Pardo, M. van Griensven and P. S. Poh, Int. J. Mol. Sci., 2019, 20, 1068 Search PubMed.
- D. Friberg, J. Bryant, W. Shannon and T. Whiteside, Clin. Diagn. Lab. Immunol., 1994, 1, 261 Search PubMed.
- M. Seitz, M. Zwicker and B. Wider, J. Rheumatol., 2001, 28, 496 Search PubMed.
- E. Mariani, G. Lisignoli, R. M. Borzì and L. Pulsatelli, Int. J. Mol. Sci., 2019, 20, 636 Search PubMed.
- R. Sridharan, A. R. Cameron, D. J. Kelly, C. J. Kearney and F. J. O'Brien, Mater. Today, 2015, 18, 313 Search PubMed.
- N. Fujiwara and K. Kobayashi, Curr. Drug Targets: Inflammation Allergy, 2005, 4, 281 Search PubMed.
- S. Li, Y. Wang, L. Guan and M. Ji, Mol. Med. Rep., 2015, 12, 4320 Search PubMed.
- C. Zhou, B. Yang, Y. Tian, H. Jiao, W. Zheng, J. Wang and F. Guan, Cell. Immunol., 2011, 272, 33 Search PubMed.
- P.-M. Chen, M.-L. Yen, K.-J. Liu, H.-K. Sytwu and B.-L. Yen, J. Biomed. Sci., 2011, 18, 49 Search PubMed.
- D. Baksh, R. Yao and R. S. Tuan, Stem Cells, 2007, 25, 1384 Search PubMed.
- R. Sarugaser, D. Lickorish, D. Baksh, M. M. Hosseini and J. E. Davies, Stem Cells, 2005, 23, 220 Search PubMed.
- L. Penolazzi, E. Lambertini, E. Tavanti, E. Torreggiani, F. Vesce, R. Gambari and R. Piva, Cell Biol. Int., 2008, 32, 320 Search PubMed.
- L. N. Handly, A. Pilko and R. Wollman, eLife, 2015, 4, e09652 Search PubMed.
- Y. Mu, L. Yang, C. Li and W. Qing, in Osteogenesis and Bone Regeneration, ed. H. Yang, Intech, 2018, ch. 4, p. 1 Search PubMed.
- M. Waters, P. VandeVord and M. Van Dyke, Acta Biomater., 2018, 66, 213 Search PubMed.
- L. Cassetta, E. Cassol and G. Poli, Sci. World J., 2011, 11, 2391 Search PubMed.
- X. T. He, X. Li, Y. Yin, R. X. Wu, X. Y. Xu and F. M. Chen, J. Cell. Mol. Med., 2018, 22, 1302 Search PubMed.
- F. Witte, H. Ulrich, M. Rudert and E. Willbold, J. Biomed. Mater. Res., Part A, 2007, 81, 748 Search PubMed.
- Q. Peng, K. Li, Z. Han, E. Wang, Z. Xu, R. Liu and Y. Tian, J. Biomed. Mater. Res., Part A, 2013, 101, 1898 Search PubMed.
- J. Sugimoto, A. M. Romani, A. M. Valentin-Torres, A. A. Luciano, C. M. R. Kitchen, N. Funderburg, S. Mesiano and H. B. Bernstein, J. Immunol., 2012, 188, 6338 Search PubMed.
- N.-Y. Su, T.-C. Peng, P.-S. Tsai and C.-J. Huang, J. Surg. Res., 2013, 185, 726 Search PubMed.
- J. Adams and J. Mitchell, Thromb. Haemostasis, 1979, 42, 603 Search PubMed.
- L. Jahangiri, M. Kesmati and H. Najafzadeh, Eur. Rev. Med. Pharmacol. Sci., 2013, 17, 2706 Search PubMed.
- L. Sun, X. Li, M. Xu, F. Yang, W. Wang and X. Niu, Regener. Biomater., 2020, 7(4), 391–401 Search PubMed.
- B. Li, H. Cao, Y. Zhao, M. Cheng, H. Qin, T. Cheng, Y. Hu, X. Zhang and X. Liu, Sci. Rep., 2017, 7, 42707 Search PubMed.
- K. Le Blanc, Cytotherapy, 2006, 8, 559 Search PubMed.
- Q. Z. Zhang, W. R. Su, S. H. Shi, P. Wilder-Smith, A. P. Xiang, A. Wong, A. L. Nguyen, C. W. Kwon and A. D. Le, Stem Cells, 2010, 28, 1856 Search PubMed.
- R. S. Waterman, S. L. Tomchuck, S. L. Henkle and A. M. Betancourt, PLoS One, 2010, 5, e10088 Search PubMed.
- P. Batten, P. Sarathchandra, J. W. Antoniw, S. S. Tay, M. W. Lowdell, P. M. Taylor and M. H. Yacoub, Tissue Eng., 2006, 12, 2263 Search PubMed.
- D. J. Prockop, Stem Cells, 2013, 31, 2042 Search PubMed.
- E. Andreeva, P. Bobyleva, A. Gornostaeva and L. Buravkova, Cytotherapy, 2017, 19, 1152 Search PubMed.
- A. Kochegarov and L. F. Lemanski, J. Stem Cells Regen. Med., 2016, 12, 61 Search PubMed.
- F. da Silva Lima, A. B. da Rocha Romero, A. Hastreiter, A. Nogueira-Pedro, E. Makiyama, C. Colli and R. A. Fock, J. Nutr. Biochem., 2018, 55, 200 Search PubMed.
- L. Wang, Y. Li, X. Chen, J. Chen, S. C. Gautam, Y. Xu and M. Chopp, Hematology, 2002, 7, 113 Search PubMed.
- A. L. Ponte, E. Marais, N. Gallay, A. Langonne, B. Delorme, O. Herault, P. Charbord and J. Domenech, Stem Cells, 2007, 25, 1737 Search PubMed.
- Z. Xia and J. T. Triffitt, Biomed. Mater., 2006, 1, R1 Search PubMed.
- P. M. Henson, J. Immunol., 1971, 107, 1535 Search PubMed.
- P. M. Henson, J. Immunol., 1971, 107, 1547 Search PubMed.
- D. Lacey, P. Simmons, S. Graves and J. Hamilton, Osteoarthr. Cartil., 2009, 17, 735 Search PubMed.
- L. Postiglione, G. Di Domenico, S. Montagnani, G. Di Spigna, S. Salzano, C. Castaldo, L. Ramaglia, L. Sbordone and G. Rossi, Calcif. Tissue Int., 2003, 72, 85 Search PubMed.
- X. Lin, Y. Zhang, J. Dong, X. Zhu, M. Ye, J. Shi, J. Lu, Q. Di, J. Shi and W. Liu, NeuroReport, 2007, 18, 1113 Search PubMed.
- S. Yoshizawa, A. Brown, A. Barchowsky and C. Sfeir, Acta Biomater., 2014, 10, 2834 Search PubMed.
- Z. Wu, T. Tang, H. Guo, S. Tang, Y. Niu, J. Zhang, W. Zhang, R. Ma, J. Su and C. Liu, Colloids Surf., B, 2014, 120, 38 Search PubMed.
- A. Chaya, S. Yoshizawa, K. Verdelis, N. Myers, B. J. Costello, D.-T. Chou, S. Pal, S. Maiti, P. N. Kumta and C. Sfeir, Acta Biomater., 2015, 18, 262 Search PubMed.
- L. Goers, P. Freemont and K. M. Polizzi, J. R. Soc., Interface, 2014, 11, 20140065 Search PubMed.
- B. Luthringer, F. Ali, H. Akaichi, F. Feyerabend, T. Ebel and R. Willumeit, J. Mater. Sci.: Mater. Med., 2013, 24, 2337 Search PubMed.
- L. Zhao, P. Abdollah, S. Do, C. Nye and B. Hantash, J. Clin. Cell. Immunol., 2013, 9, 005 Search PubMed.
- P. Lertkiatmongkol, D. Liao, H. Mei, Y. Hu and P. J. Newman, Curr. Opin. Hematol., 2016, 23, 253 Search PubMed.
- L. L. Wu, F. Feyerabend, A. F. Schilling, R. Willumeit-Romer and B. J. C. Luthringer, Acta Biomater., 2015, 27, 294 Search PubMed.
- A. Aldahmash, M. Haack-Sørensen, M. Al-Nbaheen, L. Harkness, B. M. Abdallah and M. Kassem, Stem Cell Rev. Rep., 2011, 7, 860 Search PubMed.
- K. Tateishi, W. Ando, C. Higuchi, D. Hart, J. Hashimoto, K. Nakata, H. Yoshikawa and N. Nakamura, Cell Transplant., 2008, 17, 549 Search PubMed.
- E. M. Horwitz, P. L. Gordon, W. K. Koo, J. C. Marx, M. D. Neel, R. Y. McNall, L. Muul and T. Hofmann, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8932 Search PubMed.
- A. Heiskanen, T. Satomaa, S. Tiitinen, A. Laitinen, S. Mannelin, U. Impola, M. Mikkola, C. Olsson, H. Miller-Podraza and M. Blomqvist, Stem Cells, 2007, 25, 197 Search PubMed.
- M. Sundin, O. Ringdén, B. Sundberg, S. Nava, C. Götherström and K. Le Blanc, Haematologica, 2007, 92, 1208 Search PubMed.
- K. V. Honn, J. A. Singley and W. Chavin, Proc. Soc. Exp. Biol. Med., 1975, 149, 344 Search PubMed.
- D. A. Chistiakov, M. C. Killingsworth, V. A. Myasoedova, A. N. Orekhov and Y. V. Bobryshev, Lab. Invest., 2017, 97, 4 Search PubMed.
- T. Rőszer, Mediators Inflammation, 2015, 2015, 816460 Search PubMed.
- A. Grada, M. Otero-Vinas, F. Prieto-Castrillo, Z. Obagi and V. Falanga, J. Invest. Dermatol., 2017, 137, e11 Search PubMed.
- S. Kinoshita, K. Uzu, K. Nakano, M. Shimizu, T. Takahashi and M. Matsui, J. Med. Chem., 1971, 14, 103 Search PubMed.
- M. M. Cohen and M. W. Shaw, J. Cell Biol., 1964, 23, 386 Search PubMed.
- O. Frankfurt, J. Histochem. Cytochem., 1980, 28, 663 Search PubMed.
- M. Weiss, Y. López and K. McIntosh, in Human Fetal Tissue Transplantation, ed. N. Bhattacharya and P. Stubblefield, Springer, London, 2013, p. 87 Search PubMed.
- S. Jyothi Prasanna and V. Sowmya Jahnavi, Open Tissue Eng. Regener. Med. J., 2011, 4, 28 Search PubMed.
- M. N. Walter, K. T. Wright, H. R. Fuller, S. MacNeil and W. E. B. Johnson, Exp. Cell Res., 2010, 316, 1271 Search PubMed.
- S. A. Park, C. H. Ryu, S. M. Kim, J. Y. Lim, S. I. Park, C. H. Jeong, J. Jun, J. H. Oh, S. H. Park and W. Oh, Int. J. Oncol., 2011, 38, 97 Search PubMed.
- D. E. H. Heinemann, H. Siggelkow, L. M. Ponce, V. Viereck, K. G. Wiese and J. H. Peters, Immunobiology, 2000, 202, 68 Search PubMed.
- L. Claes, S. Recknagel and A. Ignatius, Nat. Rev. Rheumatol., 2012, 8, 133 Search PubMed.
- S. Franz, S. Rammelt, D. Scharnweber and J. C. Simon, Biomaterials, 2011, 32, 6692 Search PubMed.
- R. B. Naqvi, Y. F. Joya and M. R. A. Karim, Key Eng. Mater., 2018, 778, 306 Search PubMed.
- Y. C. Li, M. H. Li, W. Y. Hu, P. D. Hodgson and C. E. Wen, Mater. Sci. Forum, 2010, 654–656, 2192–2195 Search PubMed.
- S. Agarwal, J. Curtin, B. Duffy and S. Jaiswal, Mater. Sci. Eng., C, 2016, 68, 948 Search PubMed.
- A. Romani and A. Scarpa, Arch. Biochem. Biophys., 1992, 298, 1 Search PubMed.
- R. D. Grubbs and M. E. Maguire, Magnesium, 1987, 6, 113 Search PubMed.
- A. Romani, Arch. Biochem. Biophys., 2007, 458, 90 Search PubMed.
- M. S. N. Fazliah, M. Yusuf, T. Abdullah and H. Zuhailawati, Procedia Chem., 2016, 19, 75 Search PubMed.
- L. Li, J. Gao and Y. Wang, Surf. Coat. Technol., 2004, 185, 92 Search PubMed.
- Y. Yun, Z. Dong, Z. Tan and M. J. Schulz, Anal. Bioanal. Chem., 2010, 396, 3009 Search PubMed.
- H. M. Wong, K. W. Yeung, K. O. Lam, V. Tam, P. K. Chu, K. D. Luk and K. M. Cheung, Biomaterials, 2010, 31, 2084 Search PubMed.
- K. Pichler, T. Kraus, E. Martinelli, P. Sadoghi, G. Musumeci, P. J. Uggowitzer and A. M. Weinberg, Int. Orthop., 2014, 38, 881 Search PubMed.
- D. Hong, P. Saha, D.-T. Chou, B. Lee, B. E. Collins, Z. Tan, Z. Dong and P. N. Kumta, Acta Biomater., 2013, 9, 8534 Search PubMed.
- Y. Wang, X. Xie, H. Li, X. Wang, M. Zhao, E. Zhang, Y. Bai, Y. Zheng and L. Qin, Acta Biomater., 2011, 7, 3196 Search PubMed.
- A. Burmester, B. Luthringer, R. Willumeit and F. Feyerabend, Biomatter, 2014, 4, e967616 Search PubMed.
- J. Ma, N. Zhao and D. Zhu, J. Biomed. Mater. Res., Part A, 2016, 104, 347 Search PubMed.
- A. Bertolo, D. Pavlicek, A. Gemperli, M. Baur, T. Pötzel and J. Stoyanov, J. Stem Cells Regen. Med., 2017, 13, 62 Search PubMed.
- A. Mazur, J. A. Maier, E. Rock, E. Gueux, W. Nowacki and Y. Rayssiguier, Arch. Biochem. Biophys., 2007, 458, 48 Search PubMed.
- A. Mantovani, S. K. Biswas, M. R. Galdiero, A. Sica and M. Locati, J. Pathol., 2013, 229, 176 Search PubMed.
- F. Porcheray, S. Viaud, A. C. Rimaniol, C. Leone, B. Samah, N. Dereuddre-Bosquet, D. Dormont and G. Gras, Clin. Exp. Immunol., 2005, 142, 481 Search PubMed.
- X. Zheng, K. Turkowski, J. Mora, B. Brüne, W. Seeger, A. Weigert and R. Savai, Oncotarget, 2017, 8, 48436 Search PubMed.
- F. J. Van Dalen, M. H. M. E. Van Stevendaal, F. L. Fennemann, M. Verdoes and O. Ilina, Molecules, 2019, 24, 9 Search PubMed.
- C. Mills, Crit. Rev. Immunol., 2012, 32, 463 Search PubMed.
- M. E. Bernardo and W. E. Fibbe, Cell Stem Cell, 2013, 13, 392 Search PubMed.
- F. Dazzi, L. Lopes and L. Weng, Immunology, 2012, 137, 206 Search PubMed.
- A. R. R. Weiss and M. H. Dahlke, Front. Immunol., 2019, 10, 1191 Search PubMed.
- M. Wang, Q. Yuan and L. Xie, Stem Cells Int., 2018, 2018, 3057624 Search PubMed.
- T. Lawrence and C. Fong, Int. J. Biochem. Cell Biol., 2010, 42, 519 Search PubMed.
- B. J. C. Luthringer and R. Willumeit-Römer, Gene, 2016, 575, 9 Search PubMed.
- S. Yoshizawa, A. Chaya, K. Verdelis, E. A. Bilodeau and C. Sfeir, Acta Biomater., 2015, 28, 234 Search PubMed.
- P. Libako, W. Nowacki, S. Castiglioni, A. Mazur and J. A. Maier, Magnesium Res., 2016, 29, 11 Search PubMed.
- L. T. Iseri and J. H. French, Am. Heart J., 1984, 108, 188 Search PubMed.
- S. Gao, F. Mao, B. Zhang, L. Zhang, X. Zhang, M. Wang, Y. Yan, T. Yang, J. Zhang and W. Zhu, Exp. Biol. Med., 2014, 239, 366 Search PubMed.
- S. Selleri, P. Bifsha, S. Civini, C. Pacelli, M. M. Dieng, W. Lemieux, P. Jin, R. Bazin, N. Patey and F. M. Marincola, Oncotarget, 2016, 7, 30193 Search PubMed.
- Y. Zhou, Y. Yamamoto, Z. Xiao and T. Ochiya, J. Clin. Med., 2019, 8, 1025 Search PubMed.
- F. Liu, H. Qiu, M. Xue, S. Zhang, X. Zhang, J. Xu, J. Chen, Y. Yang and J. Xie, Stem Cell Res. Ther., 2019, 10, 1 Search PubMed.
- M. François, R. Romieu-Mourez, M. Li and J. Galipeau, Mol. Ther., 2012, 20, 187 Search PubMed.
- K. S. Siveen and G. Kuttan, Immunol. Lett., 2009, 123, 97 Search PubMed.
- J. Maggini, G. Mirkin, I. Bognanni, J. Holmberg, I. M. Piazzón, I. Nepomnaschy, H. Costa, C. Cañones, S. Raiden and M. Vermeulen, PLoS One, 2010, 5, e9252 Search PubMed.
- J. Frasor, A. E. Weaver, M. Pradhan and K. Mehta, Endocrinology, 2008, 149, 6272 Search PubMed.
- A. L. Russell, R. Lefavor, N. Durand, L. Glover and A. C. Zubair, Transfusion, 2018, 58, 1434 Search PubMed.
- K. Kumagai, A. Vasanji, J. A. Drazba, R. S. Butler and G. F. Muschler, J. Orthop. Res., 2008, 26, 165 Search PubMed.
- C. Colnot, S. Huang and J. Helms, Biochem. Biophys. Res. Commun., 2006, 350, 557 Search PubMed.
- L. da Silva Meirelles, A. M. Fontes, D. T. Covas and A. I. Caplan, Cytokine Growth Factor Rev., 2009, 20, 419 Search PubMed.
- M. B. Al-Shaibani, X. n. Wang, P. E. Lovat and A. M. Dickinson, in Wound Healing-New insights into Ancient Challenges, ed. V. Alexandrescu, InTech, 2016, ch. 5, p. 99 Search PubMed.
- J. J. El-Jawhari, E. Jones and P. V. Giannoudis, Injury, 2016, 47, 2399 Search PubMed.
- R. A. Lindtner, C. Castellani, S. Tangl, G. Zanoni, P. Hausbrandt, E. K. Tschegg, S. E. Stanzl-Tschegg and A.-M. Weinberg, J. Mech. Behav. Biomed. Mater., 2013, 28, 232 Search PubMed.
- S. Ragamouni, J. M. Kumar, D. Mushahary, H. Nemani and G. Pande, Acta Histochem., 2013, 115, 748 Search PubMed.
- F. Witte, V. Kaese, H. Haferkamp, E. Switzer, A. Meyer-Lindenberg, C. J. Wirth and H. Windhagen, Biomaterials, 2005, 26, 3557 Search PubMed.
- S. F. Fischerauer, T. Kraus, X. Wu, S. Tangl, E. Sorantin, A. C. Hänzi, J. F. Löffler, P. J. Uggowitzer and A. M. Weinberg, Acta Biomater., 2013, 9, 5411 Search PubMed.
- A. Hussain, K. Bessho, K. Takahashi and Y. Tabata, Tissue Eng., Part A, 2011, 18, 768 Search PubMed.
- J. M. Díaz-Tocados, C. Herencia, J. M. Martínez-Moreno, A. M. De Oca, M. E. Rodríguez-Ortiz, N. Vergara, A. Blanco, S. Steppan, Y. Almadén and M. Rodríguez, Sci. Rep., 2017, 7, 7839 Search PubMed.
- K. Ekström, O. Omar, C. Granéli, X. Wang, F. Vazirisani and P. Thomsen, PLoS One, 2013, 8, e75227 Search PubMed.
- V. Nicolaidou, M. M. Wong, A. N. Redpath, A. Ersek, D. F. Baban, L. M. Williams, A. P. Cope and N. J. Horwood, PLoS One, 2012, 7, e39871 Search PubMed.
- P. Guihard, Y. Danger, B. Brounais, E. David, R. Brion, J. Delecrin, C. D. Richards, S. Chevalier, F. Rédini and D. Heymann, Stem Cells, 2012, 30, 762 Search PubMed.
- Z. Sheikh, P. J. Brooks, O. Barzilay, N. Fine and M. Glogauer, Materials, 2015, 8, 5671 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm00834f |
| This journal is © The Royal Society of Chemistry 2020 |