
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCold atmospheric plasma induces stress granule formation via an eIF2α-dependent pathway†
Helena
Motaln
 a,
Urša
Čerček
ab,
Nina
Recek
c,
Ana
Bajc Česnik
ad,
Miran
Mozetič
c and
Boris
Rogelj
*aef
a,
Urša
Čerček
ab,
Nina
Recek
c,
Ana
Bajc Česnik
ad,
Miran
Mozetič
c and
Boris
Rogelj
*aef
aDepartment of Biotechnology, Jozef Stefan Institute, Ljubljana 1000, Slovenia. E-mail: Boris.Rogelj@ijs.si; Tel: +386 1 477 3611
bFaculty of Pharmacy, University of Ljubljana, Ljubljana 1000, Slovenia
cDepartment of Surface Engineering, Jozef Stefan Institute, Ljubljana 1000, Slovenia
dGraduate School of Biomedicine, Faculty of Medicine, University of Ljubljana, Ljubljana 1000, Slovenia
eBiomedical Research Institute BRIS, Ljubljana 1000, Slovenia
fFaculty of Chemistry and Chemical Technology, University of Ljubljana, Ljubljana 1000, Slovenia
First published on 15th September 2020
Abstract
Cold atmospheric plasma is an ionized gas that shows promise in regenerative medical treatments, yet the mechanisms underlying its effects are still poorly understood. Plasma treatment promotes cell growth or cell death depending on the cell type and exposure parameters. To date, no early cell response to plasma, such as stress granule (SG) formation has been addressed. Cytoplasmic SGs are formed as an immediate cell response to acute stress stimuli by recruitment of over 140 proteins intertwined with cytoplasmic RNAs that leads to transient suspension of protein translation. Encouraged by the plasma effects in regenerative medicine and oncology, the atmospheric pressure plasma jet with argon gas flow is being utilized to treat SH-SY5Y cells with an inducible expression of the stress granule marker G3BP1, to gain an insight into early cell response to plasma and SG formation dynamics. Plasma effectively induces SG formation in the exposed cells in a flow/time-dependent manner, with the SG assembly clearly prompted by plasma-induced oxidative stress. Plasma causes SG formation via eIF2α-signaling, which is repressed with the SG formation inhibitor ISRIB. This insight into the early cell response to plasma treatment may lead to improved therapies in regenerative medicine and cancer treatment.
1. Introduction
Cold atmospheric plasma (CAP), hereafter referred to as plasma, is an ionized gas produced by discharges in noble gases or air under atmospheric pressure.1 Though plasma treatment has shown great promise in wound healing and cancer cell killing,2–5 the exact mechanisms remain unknown. Most studies (72%) have been performed with plasma jet and argon as a working gas.2 Though the first studies focused on direct plasma treatment, in recent years indirect plasma treatments have gained importance.2 In the latter, plasma is used for the preparation of a plasma-activated medium to which cells or tissues are exposed.2,6,7 Though plasma induces physical effects, in the plasma-activated media, its chemical effects, i.e., the production of reactive oxygen nitrogen species (RONS), pervade. In cells, these may induce membrane changes, increase the production of intracellular reactive oxygen species (ROS), cause DNA double-strand breaks, and lead to apoptosis.2,6 Because of poor understanding of the signaling pathways triggered by plasma in different cells and tissues, only 7 clinical trials have been completed so far (ClinialTrials.gov).Regarding the plasma in vitro effects, a moderate plasma treatment of fibroblasts increases their S-phase progression, the secretion of the epidermal growth factor (EGF) and the transforming growth factor-β1 (TGF-β1), and the expression of phosphorylated p65 and cyclin D1 proteins, but decreases the expression of inhibitor kappa B (IκB) protein.1 Likewise, the in vivo plasma treatment also enhances the proliferation of fibroblasts,1 lymphocytes9 and endothelial cells.10 Strikingly, cancer cells seem to be more sensitive to plasma treatment than normal cells due to their higher basal level of ROS, increased expression of aquaporins and cholesterol composition of the membrane.2 Their response to plasma treatment seems to depend, inter alia, on cell survival pathways involving p53, NF-κB, JNK, and caspases.4,11–13 Overall, it is accepted that plasma treatment causes oxidative stress to cells, which when mild enough and short-lasting leads to increased cell performance (proliferation and regeneration), whereas higher or longer plasma exposures lead to uncontrollable cell death (necrosis) or stepwise programmed cell death (apoptosis).
At the cellular level, the response of a cell to various stress stimuli starts with a halt of protein translation. This is caused by stress-induced recruitment of over 140 proteins that intertwined with cytoplasmic RNAs to form stress granules (SG). These transient RNA–protein complexes represent fibrillo-granular membraneless structures, whose size and composition depend on the type and duration of the stress.14,15 As SGs contain mRNAs, translation initiation components, and proteins affecting the mRNA function, their formation is in most respects advantageous for cells, as it minimizes their energy expenditure for control over proteins and RNA stabilization during stress. Research on SG formation, has in this respect been mainly focused on SG-nucleating proteins, such as G3BP1 (RasGAP SH3 domain binding protein 1), whose overexpression induces SG assembly and eIF2α phosphorylation.15 End of stress usually leads to SG disassembly and reboot of cell metabolism and protein translation, provided the stress stimuli does not exceed the toxicity threshold.14,15 Though plasma effects have been extensively addressed in cancer and regeneration, to date, this initial cell response to plasma involving translation inhibition and SG formation has not been evaluated.
A key protein complex in the regulation of protein synthesis is the eukaryotic translation initiation factor 2 (eIF2) that is composed of five subunits (α, β, γ, δ and ε).16 In response to stress, phosphorylation of eIF2α at serine 51 converts eIF2 to a competitive inhibitor of guanine nucleotide exchange factor eIF2B, which prevents GDP/GTP exchange for eIF2-GTP-tRNAMet. This inhibits 43S pre-initiation complex formation and cap-dependent translation, which leads to SG formation.14,16 SG formation, for many, but not all forms of stress, relies on the phosphorylation of eIF2α.17 Integrative stress response inhibitor (ISRIB) – a drug-like eIF2B complex activator, can reverse the effect of eIF2α phosphorylation by promoting the formation of the active decameric form of eIF2 and stabilizing it.16 ISRIB also makes cells resistant to the effects of eIF2α phosphorylation and blocks the SG assembly. Moreover, its addition to SG-containing cells can induce rapid disassembly of SG and liberation of mRNAs into an actively translating pool.18,19
Though many effects of plasma have been demonstrated in in vitro studies,2,20,21 none of these have addressed early stress response of the cells to plasma treatment, involving stress granule (SG) formation. It is hypothesized that plasma, similar to known oxidative stress insults such as arsenite, could trigger SG formation. Should plasma trigger remain under toxicity threshold, a dynamic process of SG formation could then be reversed upon plasma stress withdrawal, leading to SG disassembly, reboot of cell metabolism and vitality. This principle of increasing the vitality of the cells by plasma treatment is being utilized in regenerative medicine, where wound healing was found to be enhanced upon plasma exposure.3,22 But SG assembly could also lead to irreversible inclusion formation, causative for the progression of degenerative diseases such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD).23,24 The mechanisms underlying the SG response to plasma could thus be imperative for understanding both the regenerative and toxicity effects of SG formation.
To provide the primary knowledge on plasma-induced SG signaling and plasma–cell interactions, we employed an argon atmospheric pressure plasma jet to treat our established SH-SY5Y cells stably expressing mScarletI-G3BP1-Myc protein and monitored them for SG assembly. Two already characterized stressors, arsenite (oxidative stress) and sorbitol (osmotic stress), were evaluated simultaneously for acute stress treatment to allow for SG characteristic comparisons, as well as to implicate the underlying mechanisms of SG response to plasma exposure. The response dependence on eIF2α was demonstrated with ISRIB inhibitor, where fine-tuning via eIF2α levels was implicated in the regulation of SG formation. Our results provide primary insight into plasma-induced SG signaling and plasma–cell interactions. Managing the regulation of SG in cells may help us to design improved plasma therapies that can be used directly or as adjuvants in regenerative medicine and oncology.
2. Experimental
2.1. Cell line maintenance
Neuroblastoma cells SH-SY5Y (ATCC® CRL-2266™) – a subline of the parental line SK-N-SH (ATCC® HTB-11™), were purchased from ATCC and utilized in this study. FlpIn G3BP1 SH-SY5Y cells were generated (see the ESI†) and cultured as a monolayer in DMEM/F12 medium 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Sigma) supplemented with 10% FBS (tetracycline-free) and penicillin–streptomycin solution in a CO2 incubator (5%) at 37 °C and 95% air humidity. When plated into 24- and 96-well plates, trypan blue exclusion assay (0.4% trypan blue solution) was used for counting them. All the experiments were conducted with the cells of passages between 19 and 30.
:1 (Sigma) supplemented with 10% FBS (tetracycline-free) and penicillin–streptomycin solution in a CO2 incubator (5%) at 37 °C and 95% air humidity. When plated into 24- and 96-well plates, trypan blue exclusion assay (0.4% trypan blue solution) was used for counting them. All the experiments were conducted with the cells of passages between 19 and 30.
2.2. Generation of FlpIn G3BP1 SH-SY5Y cells
A pcDNA6/TR vector (Thermo Fisher Scientific) was introduced into SH-SY5Y cells for high-level expression of the tetracycline repressor protein. pcDNA6/TR vector was linearized with FspI restriction enzyme (NEB), purified with PCR Clean-up (Macherey-Nagel) and used for the transfection of SH-SY5Y cells using Xfect transfection reagent (TaKaRa) according to manufacturer's instructions. For 48 h, cells were exposed to selection medium containing tetracycline-free FBS (Gibco) and blasticidine S at 5 μg mL−1 (Sigma-Aldrich). Single-cell cloning of resistant cells was performed with dilution plating, followed by cell sorting for the homogenous expression of repressor proteins. To test the efficiency of repression, cells were transfected with a plasmid containing eGFP under a Tet operator. As the resulting SH-SY5Y-TR-eGFP cells exhibited no residual eGFP expression upon the addition of 1 μg mL−1 of doxycycline (Sigma-Aldrich), the original SH-SY5Y-TR cells were selected for FlpIn SH-SY5Y-TR-FRT cell line development. To introduce the FRT recombination site into the cells, pFRT/lacZeo was linearized with ApaI (NEB) restrictase, purified with PCR Clean-up and transfected into SH-SY5Y-TR cells (Xfect, TaKaRa). Forty-eight hours later, cells were subjected to a selection medium containing tetracycline-free FBS and 300 μg mL−1 Zeocin (Thermo Fisher Scientific). They were single-cell cloned via dilution plating. Clones of SH-SY5Y-TR-FRT cells were expanded for Southern blot analysis and performance testing (pcDNA5-FRT-TO-eGFP). FlpIn SH-SY5Y-TR-FRT cells containing a single FRT insertion site were used for the generation of an inducible SH-SY5Y-TR-FRT-mScarletI-G3BP1-Myc cell line. Gene sequence of G3BP1 was produced by PCR using oligonucleotides (5′-ctccGGTACCGAGCTCGGATCCGTGAT GGAGAAGCCTAG, 3′-CTGTTCtccggagctGATGCTAGCCTGCCGTG GCGCAAGC) and SH-SY5Y cDNA as a template. All the subsequent steps involved Gibson cloning (CloneEZ PCR Cloning kit, GenScript). Myc tag was inserted into the pcDNA5-FRT/TO vector cut by Eco321 and NotI (Fast Digest, Thermo Scientific), by annealing the oligonucleotides (5′-ATCagctccggaGAACAGAAGCTGATCAGCGAAGAGGATCTGtaGC, 3′-GCGGCCGCtaCAGATCCTCTTCGCTGATCAGCTTCTGTTCtccgga gctGAT). Then mScarletI sequence (red fluorescent protein) was added to the N-terminal of Myc post cutting the pcDNA5-FRT/TO-Myc vector by BspTI and KpnI. In the end, G3BP1 sequence was inserted between Myc-tag and mScarletI via BamHI and Eco321 restriction of the vector. SH-SY5Y-TR-FRT cells were co-transfected with pcDNA5-FRT-TO-mScarletI-G3BP1-Myc and pOG44 vectors at 1:9 ratio and a stable SH-SY5Y-TR-FRT-mScarletI-G3BP1-Myc cell line was established by hygromycin (110 μg μL−1) selection.
2.3. Plasma treatment
An indirect discharge source – atmospheric plasma jet was utilized. The argon carrier gas discharge was operated in a non-sealed electrode arrangement to produce plasma as published elsewhere.25,26 The single-electrode plasma needle jet consisted of a copper wire with a diameter of 0.1 mm, inserted inside of a 50 mm long and 1.2 mm inner diameter borosilicate glass tube. The copper wire was connected to a commercial 31 kHz high voltage alternating current power supply (Conrad Electronic). The output root mean square voltage was set to 2.3 kV and the electric current flow through an electrode of 1 mA. The carrier gas argon, with 99.99% purity, was set initially to flow rates from 2–4 SLM (standard liters per minute), but 3 SLM were used in the subsequent experiments. The cells were placed at a distance of 25 mm from the plasma nozzle during the treatment. The combination impact of flow rate, incubation time after treatment and treatment duration on SG formation was analyzed in SH-SY5Y-TR-FRT-mScarletI-G3BP1-Myc cells grown in 24- and 96-well plates. Accordingly, the medium containing the cells was changed prior treatment (500 μL per 24-well plates and 100 μL per 96-well plates). Hence during plasma treatments, the cells were covered with growth medium, which was left on them for respective incubation periods (denoted for each experiment), until their fixation and processing for microscopy analyses.2.4. Induction and inhibition of stress granule (SG) formation
For the induction of SG formation, cells were exposed to sodium arsenite (Flucka analytical), sorbitol (Sigma-Aldrich) and plasma. They were plated into 24-well plates (140000 cells per well). 24 h later, the cells were treated with arsenite (0.3, 0.5 and 1 mM) and sorbitol (0.4, 0.6 and 0.8 M) for 1.5 h. Plasma effect was tested at flow rates 1–4 SLM, and at 10, 20, 40, 60 and 80 s exposure, followed by 1, 2, 4, 12 and 24 h incubation. In the subsequent experiments, cells were exposed to plasma at a flow rate of 3 SLM for 20 s, followed by 2 h incubation. A modulator of the integrative stress response – ISRIB has been shown to inhibit SG formation via the eIF2α/eIF2B pathway.27 The ISRIB (Sigma-Aldrich) inhibitor was dissolved in DMSO to a stock solution of 2 mM concentration. Cells were treated 1 h prior SG induction with 125 nM, 250 nM, 500 nM, 1 μm and 2 μM ISRIB in 500 μL of DMEM/F12 medium.
2.5. Resazurin reduction-based cell viability assay
Resazurin reduction-based assays were used to explore the effects of arsenite and plasma on the viability of SH-SY5Y-TR-FRT-mScarletI-G3BP1-Myc cells. These were seeded in 100 μL of culture media (1 × 105 cells per mL) into 96-well plates. Twenty-four hours later, they were exposed to plasma for 5, 10, 20, 40, 80 and 160 seconds at flow rates of 2 and 3 SLM, or treated with 0.15, 0.3, 0.5 and 1 mM arsenite. Argon and non-treated cells served as a control for the plasma and arsenite treatments, respectively. Metabolic activity/proliferation of the cells was assessed by the resazurin assay. Twenty μL of 0.4 mg ml−1 resazurin solution (Sigma-Aldrich) was added to each well and incubated for 2, 3, 4, 5 and 24 h prior absorbance measurements (Tecan) at 578 nm and 630 nm wavelengths. Results normalized to the absorbance of the controls were presented.2.6. Immunocytochemistry
For immunocytochemistry, the SH-SY5Y-TR-FRT-mScarletI-G3BP1-Myc cells were seeded onto coverslips into 24-well plates (140000 cells per 0.5 mL per well) and left to attach. They were exposed to plasma treatments as described previously. Upon 2 hours of incubation, the media was removed, and the cells were washed 3× with PBS and fixed in 4% paraformaldehyde for 20 min. After PBS washes, the cells were permeabilized with 0.1% Triton for 10 min and incubated in 10% FBS for 1 h. Then, they were incubated overnight with mouse anti-eIF2α (SantaCruz Biotechnology; 1:50) and rabbit anti-phosphorylated eIF2α (Novus Biologicals; 1:200) antibodies, followed by washing and 1 h staining with secondary antibodies (anti-Ms_Alexa 488, anti Rb_Alexa647, 1:5000; Cell Signaling). Before the last wash, cells were exposed to DAPI (0.1 μg mL−1) for 5 min (Thermo Fisher) and mounted with ProLong Gold (Life Technologies). Slides were analyzed by confocal microscopy (Zeiss) using ZEN software.
2.7. Western blot analysis of eIF2α phosphorylation
For western blot analyses, the SH-SY5Y-TR-FRT-mScarletI-G3BP1-Myc cells were seeded in duplicates into 24-well plate (140000 cells per well) in 500 μl of media and left to attach. They were treated with 0.125, 0.25, 0.5, 1.0 and 2.0 μM ISRIB 0.5 h prior plasma (3 SLM, 20 s) and 0.5 mM arsenite treatment. Upon 1.5 h and 2 h of incubation, respectively, total proteins were extracted from the cells using 30 μl of RIPA buffer (20 mM Tris-HCl (pH 7.4), 150 nM NaCl, 1 mM EDTA, 1% NP-40, 10% glycerol, 1 mM sodium orthovanadate, 10 mM NaF, 10 mM β-glycerophosphate) per well. Five μL of 6× Laemmli buffer were added to the samples, boiled for 8 min at 95 °C and then 15 μl of the same was loaded onto 4–12% SDS pre-cast gels (Novex Gels, Thermo Fisher Scientific). Upon semi-dry transfer (Trans-Blot Turbo Transfer System, Bio-Rad, 12 min, 25 V, 2.5 A), onto the nitrocellulose membrane, this was blocked in 3% milk TBST for 1 h and stained overnight with rabbit anti-eIF2α (Novus Biologicals; 1:1000) and mouse anti-phosphorylated eIF2α (SantaCruz Biotechnology, 1:200), followed by 1 h of incubation with the secondary antibodies anti-Ms-HRP and anti-Rb Dylight663 (Cell Signaling). The bands were detected directly (fluorescence) or with the use of Clarity™ Western Substrate (Bio-Rad).
2.8. Image and statistical analyses
Image analyses were performed using the ImageJ program using the module for SG quantification. Cells with/without stress granules were counted using a cell counter tool and then separate cells were analyzed using a SG counter plugin (number of smoothest: 30; number of smoothest after subtraction: 3; threshold: 2000; min particle size: 1; max particle size: 1000; circularity: 0.6). All the experiments were performed in duplicate and repeated three times. The data were recorded as average ± SEM. The differences among averages were tested for significance by two-way analyses of variance (ANOVA), and Dunnett's test was adopted for post hoc comparisons. P-value < 0.05 was considered statistically significant.3. Results
3.1. FlpIn SH-SY5Y-mScarletI-G3BP1-Myc cells proved suitable for SG analyses
To provide primary knowledge on plasma-induced cell/SG signaling and plasma–cell interactions, we first established a stable FlpIn SH-SY5Y-mScarletI-G3BP1-Myc cell line with an inducible expression of G3BP1 protein, indicative of SG formation, that binds also with TIA-1 protein.28 To validate our FlpIn SH-SY5Y-mScarletI-G3BP1-Myc cell model, the cells were monitored for SG formation with the use of known stressors: sodium arsenite (oxidative stress) and sorbitol (osmotic stress) (Fig. 1A and B) and proved to exhibit a dose-dependent response at published time points by exhibiting an increased concentration of mScarletI-G3BP1 fusion protein in granular structures (ESI Fig. S1A and B†). To demonstrate that these structures are indeed SGs, we stained the stressed cells for PABP, another RNA-binding protein found in SGs.29 A distinct co-localization of G3BP1 and PABP was observed in these granular structures (Fig. 1A). Since plasma effects are known to be cell and device-type dependent,30 we next optimized the plasma treatment conditions by exposing FlpIn SH-SY5Y cells to plasma at different flow rates for various duration intervals and monitored them for the formation of SGs after 24 h (ESI, Fig. S2A†), 4 h (ESI, Fig. S2B†) and 2 h (ESI, Fig. S2C†) of incubation. Due to known dynamics of SG assembly/disassembly, only sparse SGs were observed upon 24 h of incubation in the plasma-treated cells. Plasma flow rate (standard liter per minute, SLM) and duration of treatment was found to be crucial, as 4 SLM, and 60 to 80 s duration interval proved toxic to cells by changing their morphology and lowering their adhesion (less cells attached, more floating). In contrast, plasma flow rates of 2 and 3 SLM and 2 h incubation period were proved to be the best for monitoring SG formation in FlpIn SH-SY5Y-mScarletI-G3BP1-Myc cells, where peak numbers of SGs were detected 2 h after stress stimuli (plasma) (ESI, Fig. S3†), and most of the SGs disassembled upon 24 h incubation. Besides arsenite and sorbitol, plasma confirmed to induce SG formation in FlpIn SH-SY5Y-mScarletI-G3BP1-Myc cells. | ||
| Fig. 1 Plasma induces SG formation. This is regarding the cell numbers comparative to (A) arsenite and (B) sorbitol treatment, with the quantity of SG per cell similar to arsenite effect. (C) Plasma was shown to provide highest % of cells responding with SG formation at 20 s treatment duration, flow rates 2 and 3 SLM and 2 h incubation time. (D) Treatment with 3 SLM plasma doubled the effect of 2 SLM plasma upon 2 h incubation. (E) Quantification of cells with SG upon plasma (3 SLM, 20 s), sorbitol and arsenite treatment after 1 h and (F) 2 h incubation. (G) Quantification of the number of SG per individual cell, (all together 100 cells with SG were counted) upon plasma, arsenite and sorbitol treatment. Scale bar 20 μm, arrows denote SGs. *p < 0.5, **p < 0.01 and ***p < 0.001 were considered significant. Data are presented as average ± SEM. | ||
To further fine-tune the parameters of plasma-induced SG assembly in FlpIn SH-SY5Y-mScarletI-G3BP1-Myc cells, shorter incubation periods and duration intervals were tested. These confirmed 20 s duration of plasma treatment, followed by 1–2 h incubation to exhibit the best effects (Fig. 1C). Regarding the plasma flow rates, 3 SLM more than doubled the effect of 2 SLM by causing 23% of the cell population to respond to plasma treatment with SG formation upon 2 h incubation (Fig. 1D). No such effect was observed upon 1 h incubation, when only 6–8% of the cells treated with either plasma flow rate formed SG. The effects of 2 SLM (Fig. 1E) and 3 SLM (Fig. 1F) of plasma flow rates were then compared to arsenite and sorbitol SG induction effects. Plasma at flow rates of 3 SLM showed similar effectiveness to sorbitol with respect to the formation of SG in the cells, with both being only one-third as effective as arsenite treatment that induced SG formation in over 90% of the cells. Moreover, when evaluating the SG numbers in the individual cells that responded to plasma, arsenite and sorbitol stress by SG formation (Fig. 1G), plasma treatment (3 SLM, 2 h) highly resembled the arsenite one by inducing the formation of more SGs within the cells as compared to sorbitol treatment, where groups of 1–2 and 3–4 SGs per cells appeared to predominate, and groups above 10 SGs per cells found lacking.
3.2. Plasma induces SG formation via an oxidative stress pathway
Dynamics of SG formation and assembly was shown to be trigger/stress-dependent and resulted in a distinct morphology of SGs.14 Thus, the quantitative effect of plasma on SG morphology was examined in comparison with the effects of arsenite, a known oxidative stress inducer, and with sorbitol, an osmotic stress inducer as a control (Fig. 2). Relative area of all SGs, as well as relative size of a single SG per individual cell, together with the cell size, all evaluated in relation to the number of SGs per cell, were examined in cells responding to plasma (Fig. 2A–D), arsenite (Fig. 2E) and sorbitol (Fig. 2F) by using Image J-Stress granule quantification module. The area of all SGs in the cells relative to the number of SGs per cell in plasma-treated cells increased in the range of 5–10 SGs per cells similar to that in arsenite treatment, whereas the relative size of SGs per cell in plasma-treated cells remained constant as compared to a decrease observed after the arsenite treatment. As seen in the 2nd column panels, the constant size of the plasma-induced SGs appeared independent of the number of SGs present in the cell, similar to sorbitol-induced SGs, though the latter appeared halved in size. Sorbitol treatment negatively affected the relative cell size, this probably accounted for the constant size of SGs. In contrast, the size of plasma and arsenite-treated cells increased similarly relative to the number of SGs per cell (compare 3rd column panels). Altogether our data demonstrate similarities between plasma and arsenite effects on SG formation, suggesting the oxidative stress pathway as the underlying mechanism for the effects of both. | ||
| Fig. 2 Quantitative analysis of cells responding with SG formation and SG features. The relative area of all SGs per cell, the relative size of a SG per cell and the relative cell size were quantified in cell treated with plasma at (A) 2 SLM and (B) 3 SLM after 1 h incubation, and (C) 2 SLM and (D) 3 SLM after 2 h incubation; and in (E) 0.5 mM arsenite and (F) 0.6 M sorbitol both after 1.5 h incubation. Data are presented as average ± SEM. | ||
3.3. Plasma-induced SG formation in cells does not induce cell death
Since both arsenite and plasma treatments induced an increase in the relative cell size (Fig. 2) and changed cell morphology to a more rounded shape indicative of their toxicity, we further investigated the impact of arsenite and plasma on the metabolic activity of the cells after 2, 3, 4 and 5 h and cell viability per cell death after 24 h of incubation. A resazurin assay was utilized for determining the cell metabolic activity/viability upon 2 SLM (Fig. 3A) and 3 SLM (Fig. 3B) plasma treatments in comparison with the arsenite treatment (Fig. 3C). Due to the 96-well plate format of these experiments (media volume of 100 μL), 5-times lower exposure times were used, where 4 s exposure corresponded to previous 20 s exposure of cells grown in a 24-well plate (media volume, 500 μL). The treatment with the plasma flow rate of 2 SLM decreased the metabolic activity of the cells more than the treatment of the cells with a plasma flow rate of 3 SLM. Later, flow rate confirmed not to exhibit toxicity in cells, as it increased cell recovery to above 75% of the metabolic activity/viability of the control cells upon 24 h at all exposure times tested (compare Fig. 3A and B) and showed to sustain cell vitality and not drive cells towards apoptosis or necrosis as no increase in apoptotic or necrotic cell nuclei stained with Hoechst was observed between plasma-treated and non-treated cells (Fig. 3D). The metaphase cells were counted per several visual fields on the slides (at least 800 cells per tested condition) and no difference was noted in their numbers among the treated and non-treated cells. From these, we conclude that plasma used under the conditions set in our experiments does not have any effect on the cell cycle either. In contrast, arsenite at all concentrations tested was noted to increase cell metabolism up to 5 h, with all concentrations resulting after 24 h as insignificant (p < 0.01) with a 40% viability drop (Fig. 3C), accompanied by a significant increase of apoptotic and necrotic cells (Fig. 3D). To investigate the plasma non-toxic effect on SG formation, its flow rate of 3 SLM, 20 s exposure and 2 h incubation were used in the subsequent experiments. | ||
| Fig. 3 Cell viability and apoptosis/necrosis analyses of plasma and arsenite treated cells. Resazurin assay was performed with cells exposed for 2, 4, 8, 12 and 16 s to (A) 2 SLM and (B) 3 SLM flow of plasma and (C) different arsenite concentrations. Hoechst staining enabled nuclear integrity analysis (D) to determine 3 SLM plasma (left) and 0.5 mM arsenite (right) effect on cell mitosis, apoptosis and necrosis (by evaluation of morphology of Hoechst stained cell nuclei). Data are presented as average ± SEM. | ||
3.4. Plasma induces SG formation via an eIF2α-dependent pathway
The exact mechanisms, via which plasma affects cellular processes, have not yet been defined. Moreover, none of the studies so far addressed the plasma mechanism of SG formation. Linking previous knowledge on plasma RONS generation in cell media9,30 with oxidative stress induced in cells by arsenite31 that is known to be causative for SG formation via activated eIF2α pathway, we utilized ISRIB, a known integrative inhibitor of eIF2α pathway19 to investigate plasma induced SG formation. Cells were treated with various concentrations of ISRIB 1 h before either the control arsenite (0.5 mM) (Fig. 4A–D) or plasma (3 SLM, 20 s) (Fig. 4E–H) treatment. Upon 2 h incubation, ImageJ analysis demonstrated that ISRIB decreased both arsenite (Fig. 4A and B) and plasma (Fig. 4E and F) induced SG formation in a dose-dependent manner, with even the lowest 125 nM ISRIB concentration used to account for a 30% decrease in both. Inhibition of arsenite-induced SG formation by 2 μM ISRIB was only 3.8-fold (92% to 24%), whereas nearly a 7-fold decrease of SG formation was observed in plasma-treated cells (27% to 4%), which confirms the involvement of eIF2α pathway in the plasma-induced SG formation. Likewise, ISRIB in both arsenite- and plasma-treated cells decreased the number of SGs per cell (Fig. 4C and G). In the case of arsenite, the size of SGs was found to decrease along with an increase of their number (Fig. 4D – upper panel), whereas ISRIB seemed to stabilize their smaller size (Fig. 4D – lower panel). In contrast, in plasma-treated cells, the size of SGs appears independent of their number per cell (Fig. 4H – upper panel), which upon ISRIB treatment decreases markedly in relation to the number of SGs per cell (Fig. 4H – lower panel). Although this suggests the activation of oxidative stress response pathways in cells treated with plasma to occur by a similar fashion as in arsenite-treated cells, some observed differences imply on possible activation of other pathways.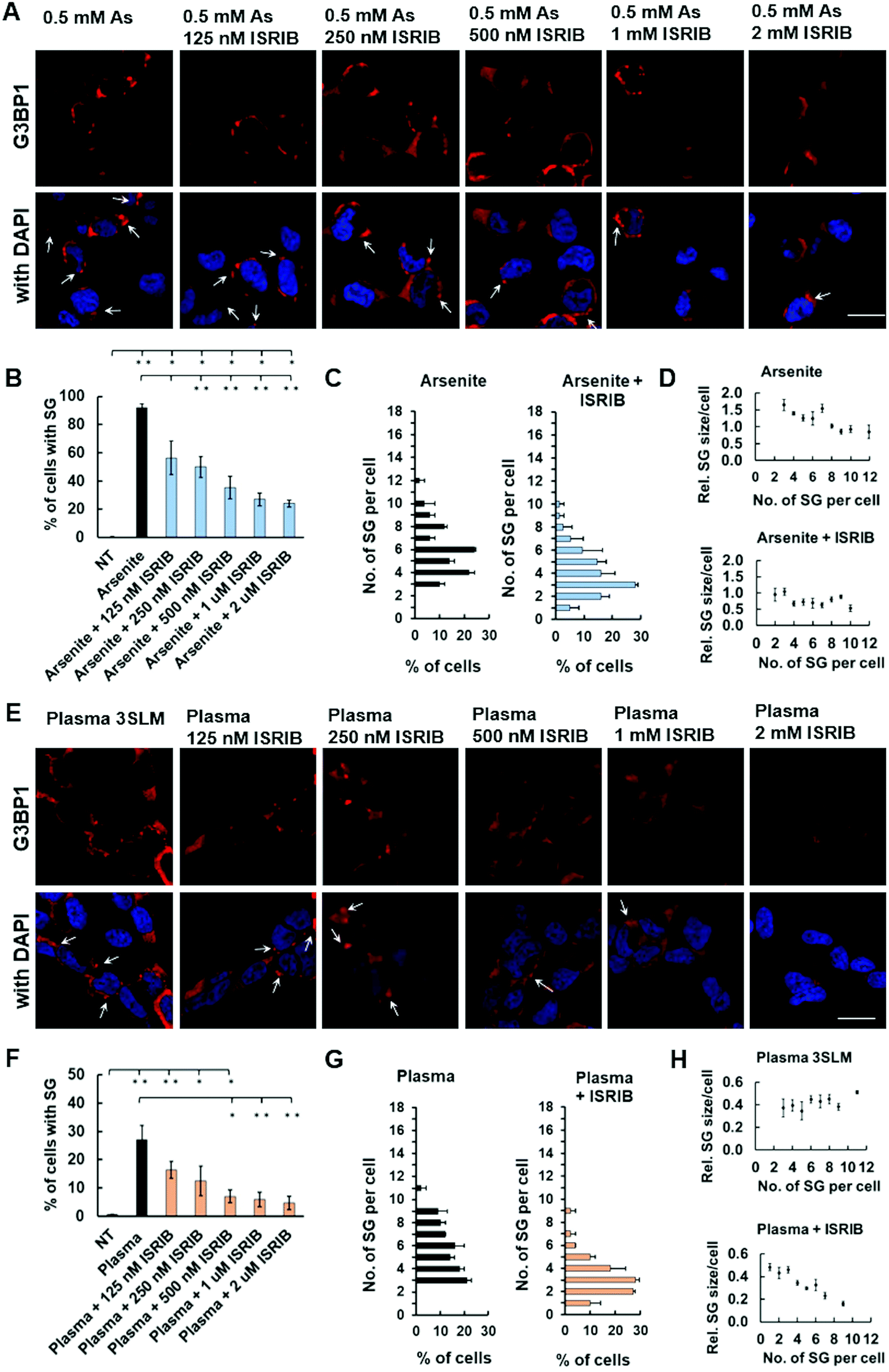 | ||
| Fig. 4 Plasma induces SG formation via eIF2α-dependent pathway. Cells were treated with 0.5 mM arsenite (A–D) or 3 SLM, 20 s plasma and ISRIB concentration ranging from 125 nM–2 μM (E–H). The % of cells with SGs (B, C, F and G) and the number of SGs per cell (D and H) was determined for each treatment by ImageJ analysis. Images were taken under 63× magnification, scale bars 20 μm, and arrows denote SGs. Data are presented as average ± SEM. | ||
3.5. ISRIB inhibition of SG formation coincides with reduced levels of phosphorylated eIF2α
As oxidative stress always results in the phosphorylation of eIF2α (p-eIF2α), quantitative assessment of the changes of eIF2α and p-eIF2α levels in ISRIB and arsenite or plasma-treated cells was performed. The selectivity of anti p-eIF2α and anti eIF2α antibodies was first tested in arsenite-treated cells (Fig. 5A), and followed by western blot analyses of total protein lysates of 0.5 mM arsenite (Fig. 5B) and plasma (3 SLM, 20 s) (Fig. 5C) treated cells, pre-treated with different concentrations of ISRIB – a modulator of the integrative stress response. | ||
| Fig. 5 ISRIB reduces the levels of p-eIF2α in stressed FlpIn SH-SY5Y cells. (A) Cells stained for p-eIF2α and eIF2α confirm arsenite induction of eIF2a pathway. Western blot analyses of cells pre-treated with 2 μM. ISRIB followed by (B) arsenite and (C) plasma treatment. Quantitative analyses performed in ImageJ (D and F) to evaluate relative amounts of p-eIF2α and eIF2α and (E and G) calculate their ratio. Scale bars 20 μm, and arrows denote SGs. Data are presented as average ± SEM. | ||
The phosphorylation of eIF2α was confirmed to occur in arsenite (Fig. 5D and E) and plasma (Fig. 5F) treated cells, where p-eIF2α was found to decrease after the ISRIB treatment in a dose-dependent manner in both. However, the relative amount of eIF2α seems to double upon arsenite treatment as compared to plasma, though p-eIF2α is found to be present in the treated cells at similar levels. Accordingly, the ratio of p-eIF2α/eIF2α decreases for 90% upon ISRIB treatment in arsenite-treated cells (Fig. 5E), whereas the p-eIF2α/eIF2α ratio decreases for only 40% (Fig. 5G) in plasma-treated ones. Together this implies on the level of eIF2α phosphorylation to play a part in the ISRIB effectiveness and the existence of alternative negative feedback loop mechanisms to fine-tune plasma-induced SG formation via the p-eIF2α pathway.
4. Discussion
Plasma-exposed eukaryotic cells in vitro demonstrate the effects such as cell detachment, cell migration/proliferation alterations, apoptosis or necrosis, all depending on cell type and exposure parameters.2,20,21 But to our knowledge, our study is the first to address plasma-induced SG formation in eukaryotic cells. For this reason, we established FlpIn-SH-SY5Y-mScarletI-G3BP1-Myc cell line with inducible G3BP1 protein expression, indicative of SG formation that can be easily monitored due to G3BP1 fusion to mScarletI red fluorescent protein. Since SG formation begins with self-oligomerization of core proteins G3BP1 (RasGAP SH3 domain binding protein 1) and TIA-1 (T-cell intracellular antigen 1),28 this very early involvement with SG makes protein G3BP1 a suitable tracker of SG assembly/disassembly dynamics. Moreover, while individual SG proteins vary widely in their dynamic properties, recent studies investigated cores with G3BP1; it was concluded that the dynamic behavior of SG cores are remarkably similar throughout their assembly.14 This makes our cell model indeed promising in providing new knowledge on SG formation, since G3BP1 expression is under inducible promoter avoiding the overproduction of the protein, that was shown otherwise to per se engage in interactions and aggregation events mediated by low-complexity sequences and internally disordered domains.14Plasma was shown to produce ROS and plenty of long-life (O3, NO, NO2 and H2O2) and short-life (OH−, O and electronically excited O) neutral particles and charged particles (ions and electrons), causing additional ROS production in the treated fluids.29,30 These ROS proved to induce cell proliferation at low concentrations and cause cell death at high concentrations.1 Presumably, ROS produced by plasma first causes the formation of long-life reactive species, which may upon moving across the cell membrane by active transport, modify ROS concentrations in the cells1 to a similar extent as these are changed during arsenite-induced oxidative stress response.33 For this reason, plasma effects on SG formation were compared with arsenite effects in our study, particularly to resolve whether the same arsenite pathway of SG formation involving the phosphorylation of eIF2α could be activated by plasma as well. Indeed, SG formation for many, but not all forms of stress, relies on the translation initiation factor eIF2α and its phosphorylation on serine 51. This prevents GDP/GTP exchange for eIF2-GTP-tRNAMet. As a result, the 43S pre-initiation complex is not generated, cap-dependent translation is inhibited and SG are produced.14 By comparing the effects of plasma and arsenite on SG formation dynamics, we were able to provide the very first explanation of plasma-induced SG dynamics, their characteristics and point to a pathway that is most likely, but not solely responsible for SG formation during plasma insult (see the scheme in Fig. 6).
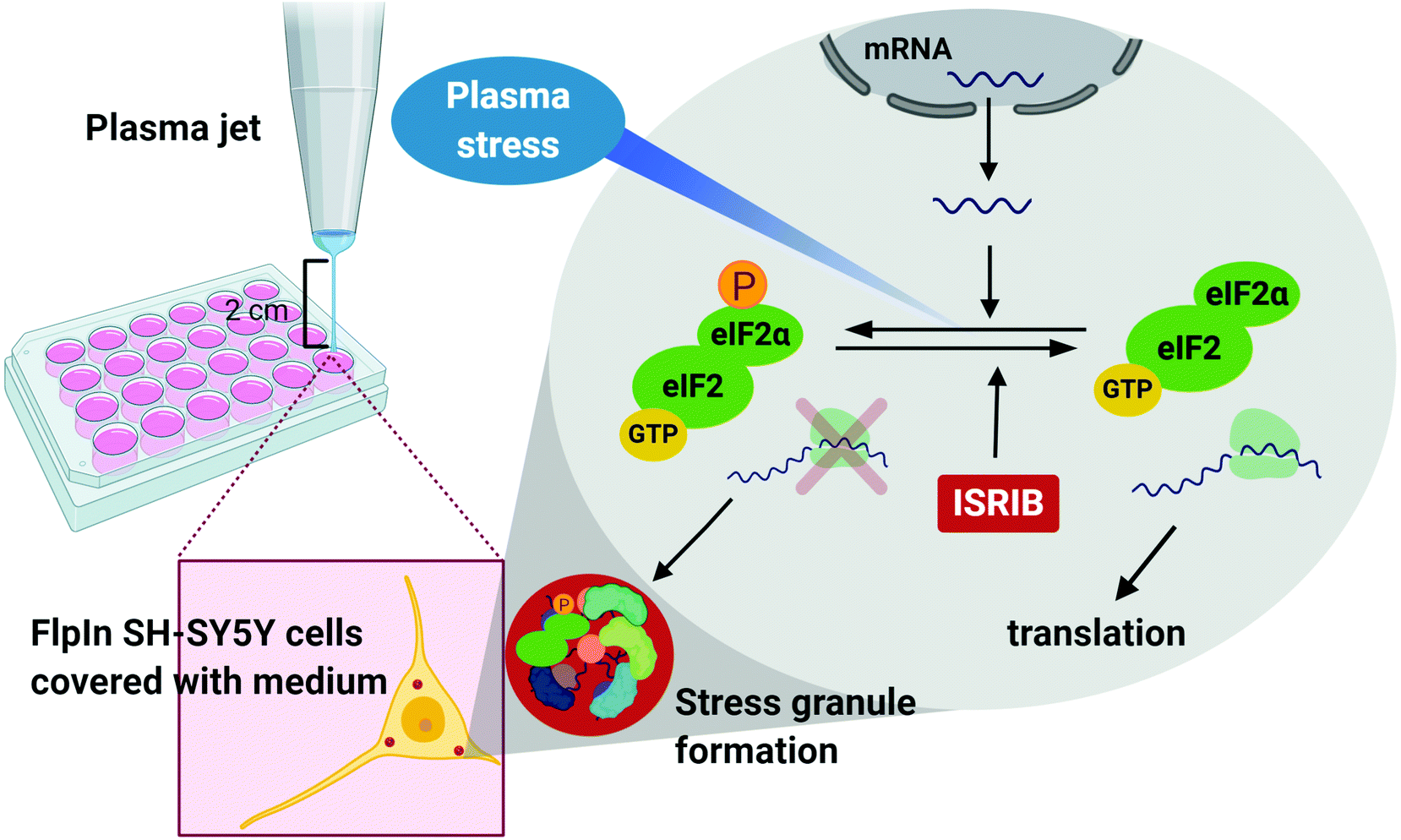 | ||
| Fig. 6 Workflow of our study where plasma jet with argon plasma flow was utilized for stress granule induction in SH-SY5Y cells that showed eIF2α signaling dependent and inhibited by ISRIB, the integrative inhibitor of stress response. Plasma induced stress granule formation via possible induction of oxidative stress response signaling (by oxidative reactive species) within the cells that involves eIF2α phosphorylation, which is inhibited by ISRIB. | ||
Since plasma effects were published to be device-type dependent,34 our treatment conditions needed to be determined experimentally. We utilized FlpIn-SH-SY5Y-mScarletI-G3BP1-Myc cells to demonstrate that plasma treatment indeed induces the formation of SG in the cells. This process is very dynamic and was shown to result in peak numbers of SGs 2 h after stress stimuli (plasma and arsenite), with most of the SGs disassembling upon 24 h incubation. The persistence of some SGs implicates also on prolonged plasma effect. Moreover, plasma was shown to induce top numbers of SGs at 20 s exposure at a flow rate of 3 SLM, which also sustained cell vitality and did not drive cells towards apoptosis or necrosis. In our experimental setup, only longer treatment times of 60–80 s caused cell detachment and massive cell death, which has already been suggested to increase the concentration of ROS above toxicity threshold,1,8,35 proving detrimental to cells in our setup. Consistently, plasma exposure times below 20 s were noted by other authors to induce proliferation of fibroblasts, whereas the ones lasting for over 30 s caused swelling of the fibroblasts and their death.1 Our viability results are also in agreement with the notion that over time cells can became resistant to increased ROS levels present below the threshold levels, and possibly ROS concentrations decreased during prolonged incubation.34,35 Yet the viability of our cells upon plasma treatment differed between the flow rates used, where 2 SLM appeared more toxic than 3 SLM. This could possibly be explained by vigorous plasma mixing with air at lower flow rates and plasma flow needed to produce long-life versus short-life reactive species1 that could underlie this differential effect.
Due to the resemblance of plasma effects4,7,21,32 to compounds33 causing ROS production and oxidative stress, we characterized plasma induced SGs in comparison with the ones induced by arsenite. Our results are indicative of oxidative stress to play, but not to have the sole role in plasma induced SG formation. Detailed analysis of SGs formed upon sorbitol, arsenite and plasma treatment revealed higher similarity of plasma induced SGs to arsenite induced ones. However, plasma (3 SLM, 2 h) did not seem to be considered as a strong inducer of oxidative stress as compared to arsenite (0.5 mM), since the percentage of cells with SGs appeared much lower after the plasma treatment. Also, the plasma induced SGs appeared smaller than the arsenite ones, implying they probably do not proceed to the SG assembly step, when several smaller SGs fuse into larger ones and is a characteristic of oxidative stress.28 The decreased strength of plasma effect is in line with recent notion that besides oxidative stress, plasma could be also triggering endoplasmic reticulum stress by protein denaturation.36
During protein synthesis, mRNAs have to pass two translational checkpoints. The first one is under the control of mechanistic target of rapamycin (mTOR) and the other one is at the assembly of the eIF2/GTP/tRNAiMet complex that is dependent of the phosphorylation of translation initiation complex eIF2α. SGs form when stressors show effect on either one of the two regulatory checkpoint units. Most often, SG formation is triggered by eIF2α phosphorylation.37 We demonstrated that similar to oxidative stress inducer – arsenite, plasma also triggers the process of SG formation via the eIF2α pathway. By using ISRIB – an inhibitor of eIF2α pathway that acts downstream of all eIF2α kinases and exclusively inhibits this pathway,19,38,39 we confirmed ISRIB to almost completely inhibit plasma induced SG formation, and to reduce arsenite induced SG formation to a much lesser extent. This is in line with the notion that ISRIB does not restore protein synthesis to 100% after saturation levels are reached, but it usually restores it to 70% only.40 Moreover, our western blot analyses confirmed the phosphorylation of eIF2α protein to occur during arsenite and plasma treatments. Although ISRIB was revealed to act downstream of eIF2α,19 we showed that in our setup, ISRIB also decreases the level of p-eIF2α in arsenite as well as in plasma treated cells. The decrease of the relative level of phosphorylated eIF2α compared to a positive control was more pronounced after sodium arsenite than plasma treatment. Consistent with that, ISRIB was shown to lower the levels of p-eIF2α in pancreatic ductal adenocarcinoma cells.31 The difference in the ISRIB inhibition level of SG formation upon arsenite and plasma exposure probably originates from the lower level of eIF2α phosphorylation. Quantitative analysis revealed lower number of cells to form SGs upon plasma treatment, which corresponds to nearly halved levels of phosphorylated eIF2α. This, together with the fact that these levels exhibit less pronounced ISRIB dose dependence, points toward the existence of putative feedback loop in the eIF2α signaling during plasma induced SG formation. By this, ISRIB though not described to inhibit p-eIF2α by direct binding, could regulate the p-eIF2α signaling.
5. Conclusions
In conclusion, we revealed that plasma treatment induces SG formation in FlpIn SH-SY5Y-mScarletI-G3BP1-Myc cells under conditions sustaining cell viability. Plasma induced SGs resemble arsenite induced SGs in nearly all characterized features. Joint characteristics of both SG types imply on plasma induced SGs to be formed via pathways triggered by oxidative species and involving eIF2α phosphorylation. The use of ISRIB – a selective inhibitor of eIF2α pathway confirmed plasma induced SG formation to be eIF2α pathway dependent. Our results provide the very first characterization of plasma induced SG. By demonstrating the levels of eIF2α phosphorylation to play a part in ISRIB effectiveness, and pointing to an alternative feedback loop in p-eIF2α pathway that could be used to fine-tune plasma induced SG formation, we provide an insight into the plasma mechanism of stress granule formation that may in future lead to improved therapies utilizing cell vitality boosting in regenerative medicine, where plasma is gaining inevitable importance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Slovenian Research Agency (grants P4-0127, J3-9263 and J3-8201).References
- X. M. Shi, G. M. Xu, G. J. Zhang, J. R. Liu, Y. M. Wu, L. G. Gao, Y. Yang, Z. S. Chang and C. W. Yao, Curr. Med. Sci., 2018, 38, 107–114 CrossRef CAS.
- A. Dubuc, P. Monsarrat, F. Virard, N. Merbahi, J. P. Sarrette, S. Laurencin-Dalicieux and S. Cousty, Ther. Adv. Med. Oncol., 2018, 10, 1758835918786475 CrossRef.
- K. Y. Cheng, Z. H. Lin, Y. P. Cheng, H. Y. Chiu, N. L. Yeh, T. K. Wu and J. S. Wu, Sci. Rep., 2018, 8, 12214 CrossRef.
- G. E. Conway, A. Casey, V. Milosavljevic, Y. Liu, O. Howe, P. J. Cullen and J. F. Curtin, Br. J. Cancer, 2016, 114, 435–443 CrossRef CAS.
- W. Li, H. Yu, D. Ding, Z. Chen, Y. Wang, S. Wang, X. Li, M. Keidar and W. Zhang, Free Radicals Biol. Med., 2018, 130, 71–81 CrossRef.
- D. Yan, W. Xu, X. Yao, L. Lin, J. H. Sherman and M. Keidar, Sci. Rep., 2018, 8, 15418 CrossRef.
- D. Boehm, C. Heslin, P. J. Cullen and P. Bourke, Sci. Rep., 2016, 6, 21464 CrossRef CAS.
- J. Y. Jang, Y. J. Hong, J. Lim, J. S. Choi, E. H. Choi, S. Kang and H. Rhim, Biomaterials, 2018, 156, 258–273 CrossRef CAS.
- X. M. Shi, G. J. Zhang, Y. K. Yuan, Y. Ma, G. M. Xu and Y. Yang, Plasma Processes Polym., 2008, 5, 482–488 CrossRef CAS.
- S. Kalghatgi, G. Friedman, A. Fridman and A. M. Clyne, Ann. Biomed. Eng., 2010, 38, 748–757 CrossRef.
- J. W. Chang, S. U. Kang, Y. S. Shin, S. J. Seo, Y. S. Kim, S. S. Yang, J. S. Lee, E. Moon, K. Lee and C. H. Kim, Sci. Rep., 2015, 5, 18208 CrossRef CAS.
- J. Köritzer, V. Boxhammer, A. Schäfer, T. Shimizu, T. G. Klämpfl, Y. F. Li, C. Welz, S. Schwenk-Zieger and G. E. Morfill, PLoS One, 2013, 8, e64498 CrossRef.
- F. Utsumi, H. Kajiyama, K. Nakamura, H. Tanaka, M. Mizuno, K. Ishikawa, H. Kondo, H. Kano, M. Hori and F. Kikkawa, PLoS One, 2013, 8, e81576 CrossRef.
- H. Mahboubi and U. Stochaj, Biochim. Biophys. Acta, Mol. Basis Dis., 2017, 1863, 884–895 CrossRef CAS.
- L. C. Reineke and J. R. Neilson, Biochem. Pharmacol., 2018, 162, 123–131 CrossRef.
- J. C. Tsai, L. E. Miller-Vedam, A. A. Anand, P. Jaishankar, H. C. Nguyen, A. R. Renslo, A. Frost and P. Walter, Science, 2018, 359, 6383 CrossRef.
- G. D. Pavitt, Wiley Interdiscip. Rev.: RNA, 2018, 9, e1491 CrossRef.
- C. Sidrauski, D. Acosta-Alvear, A. Khoutorsky, P. Vedantham, B. R. Hearn, H. Li, K. Gamache, C. M. Gallagher, K. K. Ang, C. Wilson, V. Okreglak, A. Ashkenazi, B. Hann, K. Nader, M. R. Arkin, A. R. Renslo, N. Sonenberg and K. Walter, eLife, 2013, 2, e00498 CrossRef.
- C. Sidrauski, A. M. McGeachy, N. T. Ingolia and P. Walter, eLife, 2015, 26, 4 Search PubMed.
- C. Hoffmann, C. Berganza and J. Zhang, Med. Gas. Res., 2013, 3, 21 CrossRef.
- H. Mokhtari, L. Farahmand, K. Yaserian, N. Jalili and A. K. Majidzadeh, J. Cell Physiol., 2018, 234, 6778–6782 CrossRef.
- E. Hotta, H. Hara, T. Kamiya and T. Adachi, Arch. Biochem. Biophys., 2018, 15, 644–664 Search PubMed.
- C. Vance, E. L. Scotter, A. L. Nishimura, C. Troakes, J. C. Michell, C. Kathe, H. Urwin, C. Manser, C. C. Miller, T. Hortobagyi, M. Dragunow, B. Rogelj and C. E. Shaw, Hum. Mol. Genet., 2013, 22, 2676–2688 CrossRef CAS.
- C. Vance, B. Rogelj, T. Hortobágyi, K. J. De Vos, A. L. Nishimura, J. Sreedharan, X. Hu, B. Smith, D. Ruddy, P. Wright, J. Ganesalingam, K. L. Williams, V. Tripathi, S. Al-Saraj, A. Al-Chalabi, P. N. Leigh, I. P. Blair, G. Nicholson, J. de Belleroche, J. M. Gallo, C. C. Miller and C. E. Shaw, Science, 2009, 323, 1208–1211 CrossRef CAS.
- R. Zaplotnik, Z. Kregar, M. Biscan, A. Vesel, U. Cvelbar, M. Mozetic and S. Milosevic, Europhys. Lett., 2014, 106, 25001 CrossRef.
- V. J. Law and S. D. Anghel, J. Phys. D: Appl. Phys., 2012, 45, 075202 CrossRef.
- T. Hosoi, M. Kakimoto, K. Tanaka, J. Nomura and K. Ozawa, J. Pharmacol. Sci., 2016, 131, 292–295 CrossRef CAS.
- A. Aulas, S. Stabile and C. V. Vende, Mol. Neurodegener., 2012, 7, 54 CAS.
- I. Korolov, B. Fazekas, M. Szell, L. Kemeny and K. Kutasi, J. Phys. D: Appl. Phys., 2016, 49, 035401 CrossRef.
- K. Kutasi, D. Popović, N. Krstulović and S. Milošević, Plasma Sources Sci. Technol., 2019, 28, 095010 CrossRef.
- L. R. Palam, J. Gore, K. E. Craven, J. L. Wilson and M. Korc, Cell Death Dis., 2015, 6, e1913 CrossRef CAS.
- V. S. S. K. Kondeti, C. Q. Phan, K. Wende, H. Jablonowski, U. Gangal, J. L. Granick, R. C. Hunter and P. J. Bruggeman, Free Radicals Biol. Med., 2018, 124, 275–287 CrossRef CAS.
- J. Ghosh and P. Sil, Handbook of arsenic toxicology, 2015, vol. 6, p. e1913 Search PubMed.
- E. Gjika, S. Pal-Ghosh, A. Tang, M. Kirschner, G. Tadvalkar, J. Canady, M. A. Stepp and M. Keidar, ACS Appl. Mater. Interfaces, 2018, 10, 9269–9279 CrossRef CAS.
- D. Yan, J. H. Sherman and M. Keidar, Oncotarget, 2017, 8, 15977–15995 CrossRef.
- K. Itooka, K. Takahashi, Y. Kimata and S. Izawa, Appl. Microbiol. Biotechnol., 2018, 102, 2279–2288 CrossRef CAS.
- M. Panas, P. Ivanov and P. Anderson, J. Cell Biol., 2016, 215, 313–323 CrossRef CAS.
- F. Zyryanova, F. Weis, A. Faille, A. A. Alard, A. Crespillo-Casado, Y. Sekine, H. P. Harding, F. Allen, L. Parts, C. Fromont, P. M. Fischer, A. J. Warren and D. Ron, Science, 2018, 359, 1533–1536 CrossRef.
- Y. L. Wong, L. LeBon, R. Edalji, H. B. Lim, C. Sun and C. Sidrauski, eLife, 2018, 7, e32733 CrossRef.
- M. Halliday, H. Radford, Y. Sekine, J. Moreno, N. Verity, J. le Quense, C. A. Ortori, D. A. Barrett, C. Fromont, P. M. Fischer, H. P. Harding, D. Ron and G. R. Mallucci, Cell Death Dis., 2015, 6, e1672 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S3. See DOI: 10.1039/d0bm00488j |
| This journal is © The Royal Society of Chemistry 2020 |