
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The electrospinning of a thermo-responsive polymer with peptide conjugates for phenotype support and extracellular matrix production of therapeutically relevant mammalian cells†
F. A. A.
Ruiter‡
 a,
L. E.
Sidney
b,
K. L.
Kiick
c,
J. I.
Segal
d,
C.
Alexander
a and
F. R. A. J.
Rose
*a
a,
L. E.
Sidney
b,
K. L.
Kiick
c,
J. I.
Segal
d,
C.
Alexander
a and
F. R. A. J.
Rose
*a
aSchool of Pharmacy, University of Nottingham, UK. E-mail: f.ruiter@maastrichtuniversity.nl; felicity.rose@nottingham.ac.uk; cameron.alexander@nottingham.ac.uk
bDivision of Clinical Neuroscience, University of Nottingham, UK. E-mail: laura.sidney@nottingham.sc.uk
cDepartment of Material Science and Engineering, University of Delaware, USA. E-mail: kiick@udel.edu
dFaculty of Engineering, University of Nottingham, UK. E-mail: joel.segal@nottingham.ac.uk
First published on 2nd April 2020
Abstract
Current cell expansion methods for tissue engineering and regenerative medicine applications rely on the use of enzymatic digestion passaging and 2D platforms. However, this enzymatic treatment significantly reduces cell quality, due to the destruction of important cell-surface proteins. In addition, culture in 2D results in undesired de-differentiation of the cells caused by the lack of 3D similarity to the natural extracellular matrix (ECM) environment. Research has led to the development of thermo-responsive surfaces for the continuous culture of cells. These thermo-responsive materials properties can be used to passage cells from the surface when the cell culture temperature is reduced. Here we report the development of a PLA/thermo-responsive (PDEGMA) blend 3D electrospun fibre-based scaffold to create an enzymatic-free 3D cell culture platform for the expansion of mammalian cells with the desired phenotype for clinical use. Human corneal stromal cells (hCSCs) were used as an exemplar as they have been observed to de-differentiate to an undesirable myo-fibroblastic phenotype when cultured by conventional 2D cell culture methods. Scaffolds were functionalised with a cell adherence peptide sequence GGG-YIGSR by thiol–ene chemistry to improve cell adherence and phenotype support. This was obtained by functionalising the thermo-responsive polymer with a thiol (PDEGMA/PDEGSH) by co-polymerisation. These incorporated thiols react with the norbornene acid functionalised peptide (Nor-GGG-YIGSR) under UV exposure. Presence of the thiol in the scaffold and subsequent peptide attachment on the scaffolds were confirmed by fluorescence labelling, ToF-SIMS and XPS analysis. The biocompatibility of the peptide containing scaffolds was assessed by the adhesion, proliferation and immuno-staining of hCSCs. Significant increase in hCSC adherence and proliferation was observed on the peptide containing scaffolds. Immuno-staining showed maintained expression of the desired phenotypic markers ALDH, CD34 and CD105, while showing no or low expression of the undesired phenotype marker α-SMA. This desired expression was observed to be maintained after thermo-responsive passaging and higher when cells were cultured on PLA scaffolds with 10 wt% PDEGMA/4 mol% PDEGS-Nor-GGG-YIGSR. This paper describes the fabrication and application of a first generation, biocompatible peptide conjugated thermo-responsive fibrous scaffold. The ease of fabrication, successful adherence and expansion of a therapeutically relevant cell type makes these scaffolds a promising new class of materials for the application of cell culture expansion platforms in the biomaterials and tissue engineering field.
Introduction
Current methods of adherent mammalian cell expansion for tissue engineering and regenerative medicine applications rely usually on the use of 2D culture platforms, foetal bovine serum containing media, and enzymatic digestion for cell recovery. However, 2D culture of primary cells can result in undesired de-differentiation,1–3 due to the lack of similarities to the natural extracellular matrix (ECM) environment, and enzyme passaging can significantly reduce cell quality,4–6 as a result of the destruction of important cell-surface proteins. In recent years, thermo-responsive surfaces7,8 have been used as supports for mammalian cell culture, as their changes in physicochemical properties at upper or lower critical solution temperatures (UCST, LCST) can facilitate cell detachment without the need for enzymatic passaging.9–13 PNIPAM is the most commonly used thermo-responsive polymer, however, the presence of strong hydrogen bond donors and hydrogen acceptors in its structure can lead to inter- and intra-chain association entanglement.14 By comparison, PEG-based polymers have a reversible phase transition that occurs without chain entanglement. These polymers are therefore widely used in the drug delivery and biological fields.7,10,15–17 Although, the polymer phase transitions result in measurable “hydrophilic-to-hydrophobic” behaviour changes, these differences are often only moderate in terms of their effects on cell adhesion. Accordingly, there is a need to generate materials, which provide selective functionality for specific cells to attach, while retaining their ability to release cells via a mild temperature change at the end of the cell culture and expansion period. We therefore selected a PEG-based polymer that is amenable to functionalisation without loss of the thermo-responsive behaviour, an attribute that has widely been shown in the literature.15,16,18–20Here we report the development of a new cell adhesion peptide (GGG-YIGSR) functionalised thermo-responsive fibre-scaffold based on co-electrospun poly(DL-lactide) (PLA) and poly(di(ethylene glycol) methyl ether methacrylate)-co-poly(di(ethylene-glycol) carboxylate ester ethyl thiol) (PDEGMA/SH, Fig. 1A) for enzyme-free 3D mammalian cell culture. We have exemplified this concept via the growth and recovery of human corneal stromal cells (hCSCs) in non-serum containing media. These cells were chosen as hCSCs display a well characterised phenotypic change during conventional in vitro culture from a keratocyte phenotype (quiescent, CD34+, ALDH+) to a mesenchymal stem cell/fibroblast/myofibroblast phenotype (proliferative, CD105+, α-SMA+).21 The laminin sequence YIGSR was selected for this study as previous literature has shown that interaction with this peptide sequence supports hCSCs attachment and the maintenance of a quiescent keratocyte phenotype.22
 | ||
| Fig. 1 Schematics of (A) the PDEGMA/PDEGSH (PDEGMA/SH) synthesis, (B) the formation of polymer–peptide derivative electrospun scaffolds, (C) the chemical structure of the synthesised Nor-GGG-YIGSR peptide sequence. Nor (blue block) is the norbornene acid photo-reactive group, GGG (green block) is used as a spacer and YIGSR (purple block) sequence previously identified to promote hCSCs proliferation. (D) FT-IR spectra from pure Nor-GGG-YIGSR (red), PDEGMA/SH (green) and peptide functionalised PDEGMA/PDEGS-Pep by photo-initiated thiol–ene chemistry (purple). The double bond from the norbornene acid disappeared and the amino groups of the peptide appeared on the FT-IR of the functionalised polymer–peptide. (E) Cloud point hysteresis of PDEGMA, PDEGMA/PDEGOH (PDEGMA/OH), PDEGMA/PDEGSH (PDEGMA/SH) and PDEGMA/PDEGS-Nor-GGG-YIGSR (PDEGMA/S-Pep) in water (2 mg ml−1). Arrows up are the heating spectra and arrows down are the cooling spectra. Transition temperature (Tt) shift down when polymer cools down. This is due to the aggregation of the polymer, which takes time to break up. PDEGMA/OH Tt is slightly higher in temperature due to the additional hydrophilic group OH. PDEGMA/SH polymer shows an earlier Tt change than the PDEGMA and PDEGMA/OH polymers as well as a slow steady slope when cooled. | ||
Co-spinning PLA with thermo-responsive polymers of PDEGMA and a side-chain thiol-functionalised PDEGSH generated fibrous scaffolds with ‘click’ handles for peptide coupling. The subsequent conjugation chemistries were carried out by reacting norbornene-fuctionalised peptides (Nor-GGG-YIGSR, Fig. 1C) with the side-chain thiols via a UV-mediated thiol–ene reaction (Fig. 1B). Attachment of the peptides on the scaffolds was confirmed by fluorescence labelling, ToF-SIMS and XPS analysis. The resulting bioconjugate scaffolds were assessed for their ability to support hCSCs culture by cell adhesion, proliferation and immunocytochemistry assays. The phenotypic profiling of hCSCs after thermo-responsive passaging was evaluated by flow cytometry analysis of key cell surface markers. The combined assay data indicated that the expansion of hCSCs could be conducted with high yield and retention of a therapeutically desirable phenotype without using expensive and potentially cell-damaging enzymatic methods. The ability to culture, expand and recover therapeutically relevant cell types makes these scaffolds a promising new class of materials for application in many areas of regenerative medicine, where cells are destined for the clinic.
Experimental
Polymer and peptide synthesis
General process of synthesis of norbornene functionalised peptides
Norbornene acid (Nor) functionalised peptide Nor-GGG-YIGSR was synthesised on a Rink Amide Resin (ChemPep, Wellington, FL) with a Liberty Blue™ peptide synthesizer (CEM Corporation, United Kingdom). Amino coupling cycles were as followed: Fmoc deprotection, 4× washing, coupling, coupling, 2× washing. The N-terminus of each peptide was functionalised by a norbornene acid end group using the same coupling cycle. These norbornene functionalised peptide were cleaved off the resin in a strong acid 95% trifluoracetic acid (TFA), 2.5% deionised H2O, and 2.5% triisopropylsilane under shaking for 4 h. Cleaved resin was filtered off, TFA was evaporated until 20% of the volume was left, and peptide were precipitated three times in cold ethyl. Sample was re-suspended in 80% water and 20% acetonitrile (ACN), frozen in liquid nitrogen, and lyophilised. Dried peptide was then reconstituted in the same water ACN acetic acid ratio and purified by preparative-scale reverse-phase high performance liquid chromatography (RP-HPLC) using a Waters Xbridge BEH130 Prep C-18 column. The mobile phase comprised gradients of degassed, deionised water with 0.1% TFA and ACN with 0.1% TFA, at a flow rate of 21 ml min−1. The peptide was detected by UV absorbance at 214 nm, fractions were collected and lyophilised. Molecular weight of the purified peptide was verified by electrospray ionization mass spectrometry (ESI-MS, AutospecQ, VG Analytical, Manchester, UK).Blend electrospun scaffold fabrication and polymer–peptide conjugation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF (1:1) with PLA (25% w/v) and PDEGMA/SH (10 wt%) at rt by stirring. Obtained scaffolds were fabricated using a Climate Control EC-CLI platform with rotating target collector EM RTC electrospin unit (IME Technologies, the Netherlands, Geldrop) with a 0.8 mm internal diameter (21 gauge) flat tip needle. The conducting surface was reduced to a width of 5 cm (appr. 100 cm2 surface area) by covering either side of the rotating mandrel (7.5 cm diameter) with parafilm. The climate control system was started 30 min before the run to obtain constant temperature and humidity of 20 °C and 44% respectively. A total volume of 2 ml polymer solution per run was used for each scaffold batch. Applied voltage, needle-to-collection mandrel distance and flow rate were set to 19 kV, 17.5 cm and 0.5 ml h−1 respectively.
:DMF (1:1) with PLA (25% w/v) and PDEGMA/SH (10 wt%) at rt by stirring. Obtained scaffolds were fabricated using a Climate Control EC-CLI platform with rotating target collector EM RTC electrospin unit (IME Technologies, the Netherlands, Geldrop) with a 0.8 mm internal diameter (21 gauge) flat tip needle. The conducting surface was reduced to a width of 5 cm (appr. 100 cm2 surface area) by covering either side of the rotating mandrel (7.5 cm diameter) with parafilm. The climate control system was started 30 min before the run to obtain constant temperature and humidity of 20 °C and 44% respectively. A total volume of 2 ml polymer solution per run was used for each scaffold batch. Applied voltage, needle-to-collection mandrel distance and flow rate were set to 19 kV, 17.5 cm and 0.5 ml h−1 respectively.
Electrospun scaffold characterisation
Cell culture
000 cells per cm2 in SCM and incubated at 37 °C, 5% CO2. Media was changed every 2–3 days. Passaging was performed at 80% confluency (7–9 days culture) using TripLE. Cells were used experimentally at passage 3.
000 cells per mL) in SCM and incubated at 37 °C, 2% CO2. Media was changed every 2 to 3 days culture.
Cell immunocytochemistry
Scaffolds were moved from the Cell CROWN™ and placed in a new non-treated 24 well plate, 200 μL permeabilisation buffer (0.1% Triton-X100 in PBS) was added per scaffold for immunostaining combinations B, C and D and incubated for 10 min at rt. Permeabilisation buffer was removed and scaffolds were washed with PBS (3×). All washing steps lasting 5 min each. 200 μL of blocking buffer (3% Donkey serum in wash buffer; 1% BSA, 0.3 M glycine in PBS) was added per scaffold for all immuno-staining combinations and incubated for 1 h at rt.
Subsequently, 200 μL of primary antibody solution was added according to the immuno-staining combination per scaffold at the required dilution factor prepared in wash buffer (1% BSA, 0.3 M glycine in PBS; see Table S2†). Samples were incubated at 4 °C for 16 h. Negative controls were incubated in wash buffer only. The primary antibody solutions were removed, scaffolds were rinsed with wash buffer (3×). Secondary antibodies (200 μL) were added per scaffold, including negative controls and incubated for 1 h at rt in the dark (covered in foil). Subsequently, washed with PBS (3×). Scaffolds of combination C were incubated for 20 min in the dark in 200 μL actin solution (see dilution, Table S2†) and washed with PBS (3×).
All nuclei were stained with DAPI. DAPI (200 μL) was added per scaffold and incubated for 10 min at rt in the dark, washed with PBS (3×) and stored in PBS at 4 °C.
hCSCs thermo-responsive detachment studies and phenotype expression after detachment by flow cytometry
Percentage of cells detached was calculated by subtracting the number of cells on the scaffold left after passaging with the initial number of cells before passaging. This number was then defined by initial number of cells and multiplied by 100.
Characterisation techniques
:1 ratio, SH:maleimide group), incubated for 30 min at rt and washed with PBS (3×) and imaged under confocal; images where enhanced to observed lower signals on the fibres.
:1 ratio, peptide:ATTO group), incubated for 30 min at rt and washed with PBS (3×) and imaged under confocal; images where enhanced to observed lower signals on the fibres.
Results and discussion
Polymer synthesis
The co-polymers PDEGMA/SH in different ratios (100:0, 98:2, 97:3, 96:4, and 90:10, PDEGMA to PDEGSH respectively) were synthesised by a two-step route (scheme. Fig. 1A). PDEGMA/PDEGOH polymer was synthesised by free radical synthesis with subsequent thiol functionalisation via a condensation reaction to form the PDEGMA/SH product. Structures were confirmed by 2D-NMR analysis (Fig. S1–S18†) with the disappearance of the C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2OH signal (H1-NMR; 3.7–3.8 ppm, t, 2H and C13-NMR; 61 ppm) and the appearance of the C2SH peaks (H1-NMR; 2.9 ppm, t, 2H and C13-NMR; 34 ppm) in the H1 and C13-NMR of the end product PDEGMA/SH (Fig. S1–18 for NMR and Table S3† for GPC). Percentages of free thiol in the co-polymers were confirmed by the Ellman's assay (Table 1). The phase transition temperatures (Tt) of the polymers were estimated by cloud point measurements (Fig. 1E, S19 and Table S4†). The norbornene functionalised peptide Nor-GGG-YIGSR was synthesised on a rink amide resin, purified by HPLC and its structure verified by electrospray ionisation mass spectrometry (ESI-MS, Fig. S20†). Upon reaction of the peptide with the thiolated polymer, a decrease in percentage of free thiols was observed at increasing UV exposure times (0, 5, 10 and 15 min), with a conversion of 65% after 15 min (Table 1). Fourier-transform infrared (FT-IR) analysis showed the disappearance of the norbornene double bond (1530 cm−1) and the appearance of the amino-groups (1655 cm−1) of the peptide in the case of the reacted peptide-thiol polymer (Fig. 1D). Cloud point measurements of the PDEGMA/S-Nor-GGG-YIGSR derivatised polymer showed transition temperatures (22 °C when heated and <18 °C when cooled, Fig. 1E and Table S4†), as compared with the PDEGMA/SH precursor, confirming retention of thermo-responsive behaviour after peptide-functionalisation.
2OH signal (H1-NMR; 3.7–3.8 ppm, t, 2H and C13-NMR; 61 ppm) and the appearance of the C2SH peaks (H1-NMR; 2.9 ppm, t, 2H and C13-NMR; 34 ppm) in the H1 and C13-NMR of the end product PDEGMA/SH (Fig. S1–18 for NMR and Table S3† for GPC). Percentages of free thiol in the co-polymers were confirmed by the Ellman's assay (Table 1). The phase transition temperatures (Tt) of the polymers were estimated by cloud point measurements (Fig. 1E, S19 and Table S4†). The norbornene functionalised peptide Nor-GGG-YIGSR was synthesised on a rink amide resin, purified by HPLC and its structure verified by electrospray ionisation mass spectrometry (ESI-MS, Fig. S20†). Upon reaction of the peptide with the thiolated polymer, a decrease in percentage of free thiols was observed at increasing UV exposure times (0, 5, 10 and 15 min), with a conversion of 65% after 15 min (Table 1). Fourier-transform infrared (FT-IR) analysis showed the disappearance of the norbornene double bond (1530 cm−1) and the appearance of the amino-groups (1655 cm−1) of the peptide in the case of the reacted peptide-thiol polymer (Fig. 1D). Cloud point measurements of the PDEGMA/S-Nor-GGG-YIGSR derivatised polymer showed transition temperatures (22 °C when heated and <18 °C when cooled, Fig. 1E and Table S4†), as compared with the PDEGMA/SH precursor, confirming retention of thermo-responsive behaviour after peptide-functionalisation.
| Percentage of free thiols in co-polymers | ||||
|---|---|---|---|---|
| % PDEGOH | C polymer [mmol ml−1] | Abs | C thiols [mmol ml−1] | % PDEGSH |
| 2 | 70 × 10−4 | 0.0877 | 1.2 × 10−4 | 1.7 |
| 3 | 70 × 10−4 | 0.1249 | 1.5 × 10−4 | 2.2 |
| 4 | 70 × 10−4 | 0.2483 | 2.6 × 10−4 | 3.7 |
| 10 | 70 × 10−4 | 0.7328 | 6.8 × 10−4 | 9.7 |
| Percentage of free thiols after thiol–ene chemistry | |||||
|---|---|---|---|---|---|
| UVtime [min] | C polymer [mmol ml−1] | Abs | C thiols [mmol ml−1] | % PDEGSH | % conv. |
| 0 | 70 × 10−4 | 0.2483 | 2.6 × 10−4 | 3.7 | 0 |
| 5 | 19 × 10−4 | 0.0300 | 6.8 × 10−4 | 2.0 | 46 |
| 10 | 19 × 10−4 | 0.0295 | 6.8 × 10−4 | 2.0 | 46 |
| 15 | 70 × 10−4 | 0.0537 | 0.9 × 10−4 | 1.3 | 65 |
Electrospun scaffold fabrication and characterisation
The obtained PDEGMA/SH polymers were successfully blend electrospun with PLA into uniform fibres (19 kV, 17.5 cm, 0.5 mL h−1 and 25 w/v% PLA and 10 wt% PDEGMA/SH polymer in Ac/DMF) with an average fibre diameter range of 800–1200 nm (Fig. S22 and Table S5,† optimised via previous DoE work25). XPS analysis and fluorescence labelling confirmed the presence of free thiols (S(2p) peak at 168.9 eV and fluorescein-5-maleimide labelling respectively) and peptide (N(1s) peak at 399.8 eV and ATTO NHS ester labelling respectively) on the surface of the scaffolds (Fig. S22 and Table S6†). The distributions of the free thiols and peptides on the scaffolds surface were investigated further by ToF-SIMS analysis. Mass spectrometry peaks of the ion fragments were identified and confirmed through literature (Table S7†).26–29 The ion fragment CH3O− attributed to the underivatised PDEGMA was present on the surface, and a decrease in peak intensity of CH3O− was observed for all the scaffolds containing PDEGMA/SH co-polymer (Fig. 2A). ToF-SIMS peak distribution images indicated that SH groups were in regions corresponding to the fibre positions on the substrate (Fig. 2C and Fig. S22†). An even in S− and SH− absorptions were detected for the free thiols on the fibrous surface, confirming an even thiol–ene active distribution sides. A decrease in S− and SH− absorptions were detected for the thiol–ene derivatised scaffolds (Fig. 2A). The CNO− peak of the peptide amino-groups were observed for all scaffolds incubated with peptide (Fig. 2A). Significantly higher intensity was detected for the scaffold 3 PDEGMA/S-peptide, with a decrease in CNO− intensity being observed for 4 and 10 PDEGMA/S-peptide (Fig. 2A).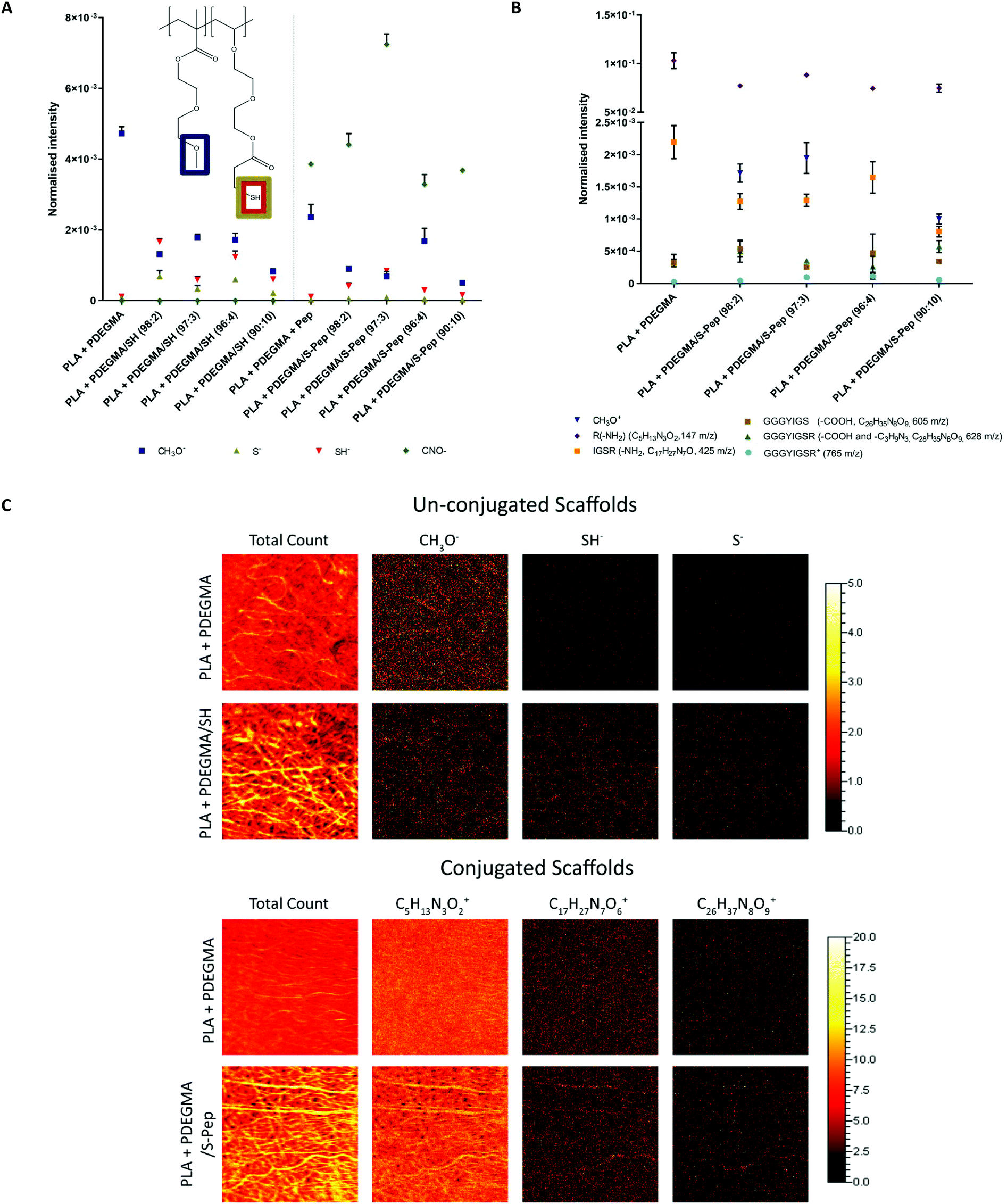 | ||
| Fig. 2 (A) Total intensity of the ToF-SIMS peaks CH3O− (31 m/z), SH− (33 m/z) and S− (32 m/z) from the free thiol containing scaffolds. (B) Total intensity of different sequences of the Nor-GGG-YIGSR peptide presences on the scaffold surfaces, (R(–NH2, 147 m/z), GSR(–COOOH, 326 m/z), IGSR(–NH2, 425 m/z), GGGYIGS(–COOH, 605 m/z), GGGYIGSR(–COOH, 628 m/z) and GGGYIGSR(765 m/z) (n = 3 measurements areas on one scaffold each, data normalised against total count, error bar = SD). (C) ToF-SIMS signal intensity images. | ||
Nor-GGG-YIGSR peptide positive ion fragment signals were identified and were corroborated to literature30,31 (see fragment structures of identified signals in Fig. S23†). Peptide fragments were detected for all scaffolds, including the PLA + PDEGMA (100:0) scaffolds (Fig. 2B). This suggested that non-specific electrostatic binding of the peptide occurred with the surface of the scaffolds. The distribution images of the signal, an even distribution of peptide fragment peaks was observed on the PLA + PDEGMA (100:0) scaffold with no thiols present, without a defined fibre morphology (Fig. 2C). However, in case of the peptide-derivatised scaffolds, more defined fibre morphologies were found (see Fig. 2C). This observation indicates the occurrence of specific reaction of the fibre surfaces when free thiols are available, as well as some, albeit low-level, non-specific peptide interaction at the surface of the fibres with no thiols present.
Cell expansion and phenotype support
hCSCs were cultured on the fabricated scaffolds for up to 9 days without loss of scaffold integrity (see protocol ESI section 1†). The media composition of DMEM/F12 with 20% serum replacement, fibroblast growth factor and leukaemia inhibitory factor (SCM) has previously reported to support the keratocyte phenotype of hCSCs23 and was therefore used in this study. Enhanced proliferation was observed on the scaffolds with an increased content of the laminin mimetic peptide sequence Nor-GGG-YIGSR compared to the PLA + PDEGMA scaffolds (Fig. 3A). Interestingly, significantly higher proliferation was also found on PLA with PDEGMA/SH in the fibres (Fig. 3A). This may be due to presence of free thiols, which promote protein interaction and therefore cell adherence. Morphology staining (actin and vimentin for the cytoskeleton) studies showed clusters of cells on PLA, and PLA with 10 wt% PDEGMA (100:0, Fig. 4 and S25–S28†), elongated cell morphologies on PLA with PDEGMA/SH (90:10, Fig. S28†), and an increase in a spindle-like morphology of the cells on the peptide derivatised PLA scaffolds with increased PDEGMA/S-Nor-GGG-YIGSR present (Fig. 4 and S26†). The round morphology of the cells on the PLA, and PLA with 10 wt% PDEGMA (100:0) scaffolds indicated a limited interaction between the cells and the fibrous scaffold. This in turn suggested that protein deposition of the media was not enhanced when the adhesion peptides were merely adsorbed (as detected by ToF-SIMS analysis) rather than being covalently bound to the fibre surface. The elongated morphologies of cells attached to the thiol-functionalised scaffolds was due most probably to direct cell surface-thiol reactions, for example via surface exposed cysteines. This assertion is supported by prior data showing that thiolated surfaces can result in improved ECM protein adsorption and subsequently enhanced cell attachment and proliferation.32–34 However, significantly more cells exhibiting a spindle-like morphology were observed when the laminin mimetic sequence YIGSR was present (see Fig. 4). Uzunalli et al.22 reported a similar morphology of hCSCs when cultured on YIGSR-containing nanofibres compared to fibronectin-mimetic RGD-peptide nanofibres, as well as increased proliferation, supporting the findings reported here.
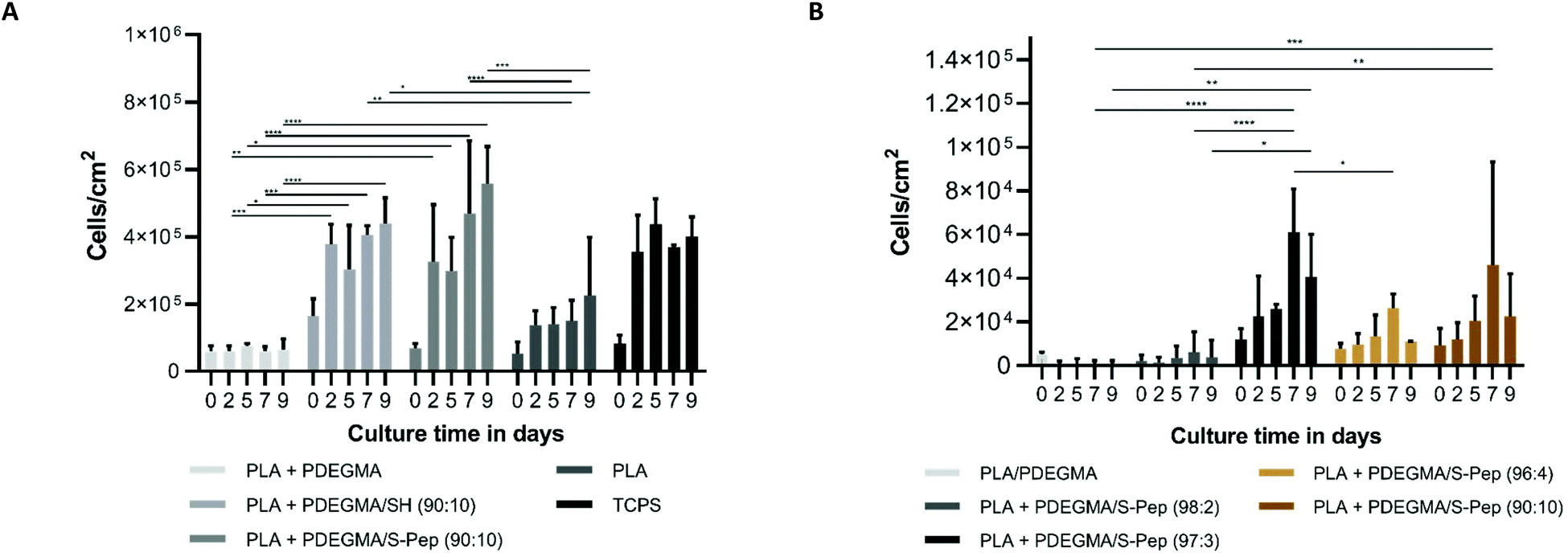 | ||
| Fig. 3 (A) Cell proliferation in cells per cm2 of the scaffold different scaffolds types compared to control TCPS over 9 days of culture. (B) Cell proliferation in cells per cm2 of the peptide derivatised scaffolds with different concentration of peptide attached over 9 days of culture (n = 3 scaffolds, two-way ANOVA, * = <0.05, ** = <0.01, *** = <0.001 and **** = <0.0001, error bars = SD). | ||
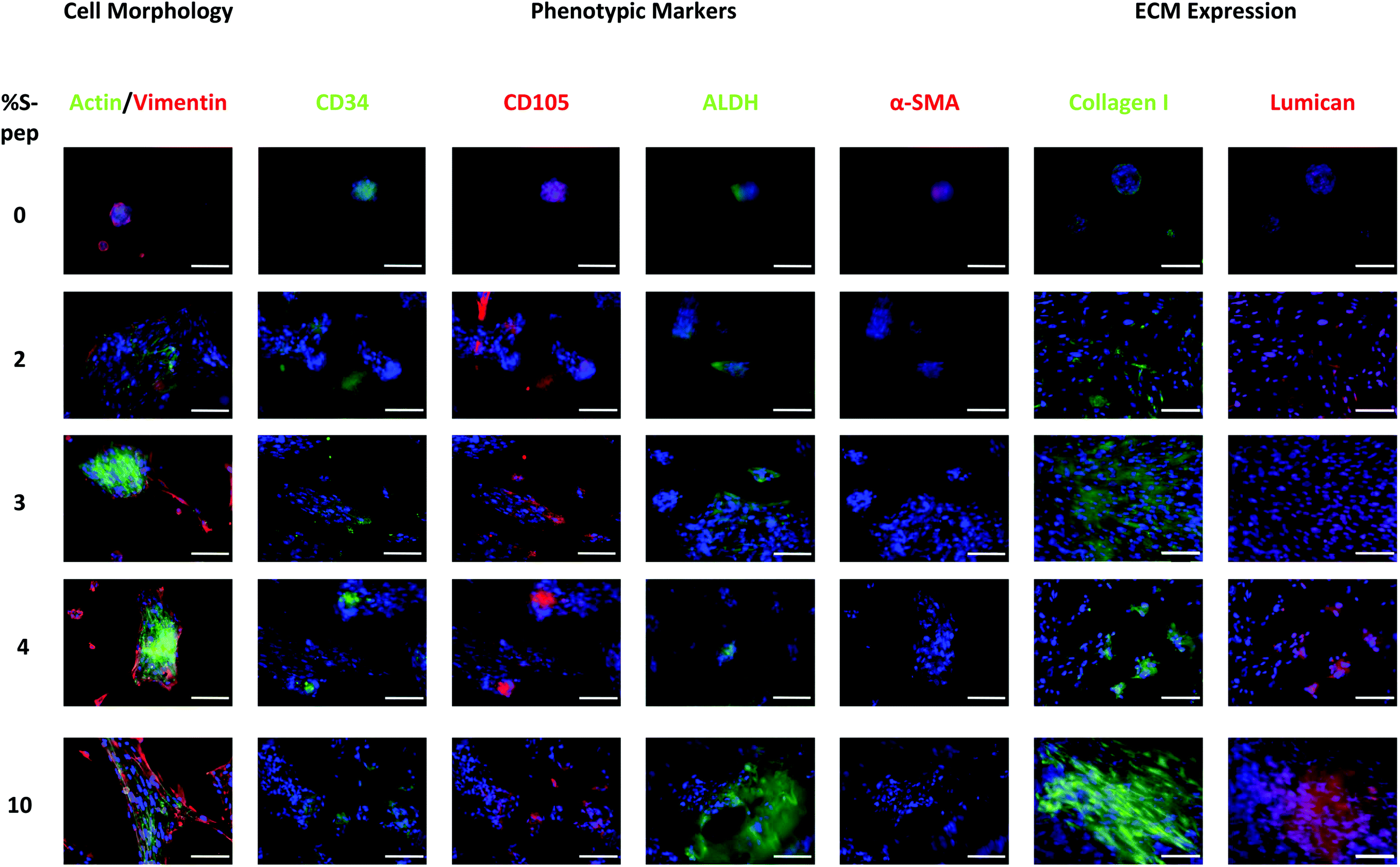 | ||
| Fig. 4 Immuno-staining to observe hCSC morphology, phenotype expression and ECM expression when cultured on the peptide derivatised PDEGMA/PDEGS-Pep scaffolds at different percentage of peptide. Nuclei are stained with DAPI. Images are enhanced to achieve higher resolution images and separated channel images can be found in ESI Fig. S25–27.† Scale bars correspond to 100 μm. | ||
Immuno-staining of the hCSCs showed the quiescent keratocyte phenotypic markers CD34 and ALDH, and mesenchymal stem cell (MSC) expression of CD105 on all the different scaffolds (see Fig. 4 and Fig. S26 and 29†). In addition, the expression of myofibroblast marker α-SMA was found when cells were cultured on the PLA and PLA with 10 wt% PDEGMA (100:0) scaffolds, and a very low expression of α-SMA was also observed when cells were cultured on the PLA fibres with 10 wt% PDEGMA/SH (90:10, Fig. S26 and 29†). This indicates that although the scaffolds with free thiols available do support cell adherence and proliferation as observed in Fig. 3A, α-SMA was still expressed indicating the expression of a myofibroblast phenotype. On the other hand, no α-SMA expression was detected when cells were cultured on any of the PLA fibres functionalised with 10 wt% PDEGMA/S-Nor-GGG-YIGSR (90:10). These data confirmed that the peptide supported the quiescent keratocyte phenotype and suppressed de-differentiation of the keratocytes to their activated myofibroblast phenotype.
ECM expression was investigated to confirm the hypotheses that the free thiol group-containing scaffold supported ECM expression and adsorption and hence cell adhesion and proliferation as observed in Fig. S28.† Collagen I expression was observed throughout all the scaffold types (Fig. 4 and S27†), however, this protein increased in abundance and was more evenly distributed on the peptide containing scaffolds (Fig. 4 and S27†). While, collagen I expression was observed near to the clusters of cells on the PLA + PDGEMA (100:0) scaffold with absorbed peptide only (Fig. 4 and S27†). These results confirmed that PDEGMA/S-Nor-GGG-YIGSR peptide functionalisation on the scaffold led to an increased expression of collagen I.
Stromal keratocytes express the keratan sulphate proteoglycan, lumican, a corneal transparency factor in a normal undamaged cornea.32,33 Lumican interacts with collagen fibrils to maintain spacing and transparency in a healthy cornea.34 Production is significantly reduced during wound healing and phenotypic change to a fibroblast morphology, and thus the presence of lumican can be considered a marker of the quiescent phenotype of hCSCs. As apparent from Fig. 4, lumican expression was found on the scaffolds with peptide present; including the PLA + PDEGMA (100:0) scaffolds with non-specific bound peptide. No lumican was present on the scaffold with free thiols, without peptide present (see Fig. S27†). This indicates that the presence of peptide at the surface of the fibres supported ECM expression, while free thiols at the surface do not. However, myofibroblast differentiation and clusters of cells were still observed on the PLA + PDEGMA (100:0) scaffolds, suggesting that the non-specifically bound peptide did not support strong cell interactions with the scaffold surface. It was observed that cells spread out across the surfaces only when the peptide was covalently bound to the fibres. Increased lumican expression was detected for the peptide bound scaffolds with 3, 4 and 10 mol% PDEGMA/S-peptide attached, providing support for assertion (Fig. 4). As the derivatised scaffolds with the Nor-GGG-YIGSR peptide were observed to promote the desired quiescent phenotype, spindle-like morphology and ECM expression, the cellular responses on the surfaces with different percentages of peptides attached were investigated. After 9 days cell culture, significantly higher proliferation was observed for 3 and 10% peptide-derivatised scaffolds (97:3 and 90:10, Fig. 3B). The expression of the quiescent keratocyte phenotype CD34 and ALDH markers were detected for all different concentrations of peptide (Fig. 4). A significantly increased expression of ALDH was observed for the 3 mol% peptide scaffold. As ALDH expression plays an important role in the maintenance of corneal transparency, this indicated a desirable response of the cells in term of their culture on the derivatised fibres. In addition, CD105 MSC marker expression was found on all scaffolds, suggestive of a partial loss of the differentiated quiescent phenotype to a more stem cell-like phenotype.24 Myofibroblastic α-SMA expression was observed for those cells cultured on scaffolds containing 2 mol% peptide (98:2, PDEGMA/S-pep) and on the 0% control (100:0), indicating a threshold of peptide content needed to avoid myofibroblast activation. Peptide concentrations of 2 mol% were too low to support the quiescent phenotype and resulted in a myo-fibroblastic change, while cells cultured on scaffolds containing 3, 4 and 10 mol% peptide generated no α-SMA expression. The previously rounded and cluster cell morphology was observed on the scaffolds containing 0 and 2 mol% peptide concentrations, while a more fusiform morphology was observed when cultured on scaffolds with peptide-concentrations 3, 4 or 10 mol% (Fig. 4). Moreover, lumican ECM expression was detected when cells were cultured on scaffolds with higher percentages of peptide on the surface (3, 4, and 10 mol%). It can therefore be concluded that scaffold fibres with more than 2 mol% of Nor-GGG-YIGSR covalently bound to their surfaces significantly improved hCSCs attachment, proliferation, ECM expression and supported a quiescent cell phenotype.
Enzymatic-free passaging
The PLA with thermo-responsive PDEGMA/S-Nor-GGG-YIGSR polymer scaffolds were not only designed to improve cell attachment and phenotype support, but also to prevent cell damage, which can occur through enzymatic passaging. Many studies to date have investigated the potential of the thermo-responsive polymer, PNIPAM, for corneal cell type detachment.8,35–37 We have recently shown that blend electrospinning of thermo-responsive polymer poly(PEGMA188) with a bulk polymer of choice (amenable to electrospinning) will render the fibres thermo-responsive. Mammalian cell adhesion, viability, proliferation and phenotype on this fibrous culture system over numerous thermal enzyme-free passages was achieved.38 However, no other studies are reported to date utilising such thermo-responsive scaffolds for human primary cell culture. A preliminary experiment illustrating the ability to culture and detach cells from the PLA + PDEGMA and PLA + PDEGMA/S-Nor-GGG-YIGSR-containing scaffolds, and the subsequent quantitative assessment of the cell phenotype by flow cytometry analysis (FACS), was performed. hCSCs grown on peptide-derivatised fibrous scaffolds containing 10 wt% of different molar ratios of PDEGMA/S-pep were passaged after 9 days of cell culture at 37 °C by placing the scaffolds in the fridge for 30 min at 4 °C (section 1 ESI†). Cell detachment was observed from all scaffolds (Fig. 5A). This demonstrates the thermo-responsive passaging capability of the scaffold containing peptide on the surface. However, fluctuating percentages of cells detached were observed from some of the 2 and 10 mol% PDEGS-Nor-GGG-YIGSR (98:2, 90:10) containing scaffolds (Fig. 5A). This may have been due to heterogeneous cell attachment to the lowest percentage peptide derivatives (PDEGMA/S-Nor-GGG-YIGSR (98:2)) and very strong adhesion to the more extensively peptide-functionalised fibres (PDEGMA/S-Nor-GGG-YIGSR (90:10)). Nonetheless, more consistent cell detachment was observed from the scaffolds containing 3 and 4 mol% PDEGS-Nor-GGG-YIGSR (97:3 and 96:4). The presence of extracellular matrix after detachment was not investigated in this work and will be included in future work.
 | ||
| Fig. 5 (A) Percentage of cells detached after thermo-responsive passaging of the different concentrations of peptide on the surface (n = 3 scaffolds, two-way ANOVA, error bars = SD). (B) Flow cytometry percentage of cell population expressing the desired keratocyte marker CD34 and/or mesenchymal stem cell marker CD105 after thermo-response detachment from the different scaffolds (n = 3 cells detached from 3 scaffolds per scaffold type, control cell passage with trypsin from TCPS). (C) Flow cytometry percentage of cell population expressing the desired keratocyte marker ALDH and/or the undesired myo-fibroblastic marker α-SMA after thermo-response detachment from the different scaffolds (n = 3 cells detached from 3 scaffolds per scaffold type, control cell passage with trypsin from TCPS). (D) Combined percentage of cell population that expresses α-SMA on the different scaffolds (n = 3 cells detached from 3 scaffolds per scaffold type, control cell passage with trypsin from TCPS, * = <0.05). | ||
Flow cytometry analysis of cell surface markers of cells detached from the scaffolds was carried out to validate immuno-staining findings. Cells cultured on 3 mol% peptide scaffolds were observed to have an increased sub-population of 16% of cells that only expressed the CD34+/CD105− keratocyte marker (Fig. 5B and Tables S8–9†) compared to those cultured on all other scaffolds. However, CD34−/CD105+ and CD34+/CD105+ sub-populations combined constituted half of the total cell population (53%, Table S8†) whereas CD34+/CD105+ and CD34+/CD105− cells comprised 45% (Table S8†) of the total cell population. Therefore, a significant percentage of the population exhibited MSC characteristics besides those of the desired keratocyte CD34 marker. A similar sub-population percentage of CD34−/CD105+ and CD34+/CD105+ cells (51%, Table S8†) was detected when cultured on the scaffolds with 4 mol% peptide on the surface, nevertheless there was an increase in the proportion of cells which expressed the CD34 marker (38% of total cell population, Table S8†). Significantly less CD34+/CD105+ expression was observed for all scaffolds compared to the 2D TCPS control when comparing the Y-mean (Table S9†). This observation shows that the 3D culture environment significantly reduced the expression of the MSC marker CD105, while no significant difference was seen for the different molar percentage content of peptides (Tables S8 and S10–11†). In addition, an increased expression of ALDH+/α-SMA− was demonstrated when cells were cultured on 3 and 4 mol% peptide scaffolds (Fig. 5C). In terms of the total cell population, 24, 28, 19 and 32% (ALDH−/α-SMA+ and ALDH+/α-SMA+ combined, Table S8†) for 2, 3, 4 and 10 mol% peptide-functionalised fibres, respectively, were shown to express the α-SMA activated myofibroblastic marker. This was significantly less than the 93% and 43% observed for the 2D control and PLA + PDEGMA scaffold respectively. Hence, the 3D structure of the scaffolds containing covalently bound peptide on the surface reduced the differentiation to the undesired activated myo-fibroblastic phenotype compared to the 2D control. Although no significant difference could be observed between the scaffolds for CD34/CD105 expression, a significant decrease of ALDH−/α-SMA+ was seen for the scaffolds with 3 and 4 mol% peptide compared to 2D and the adsorbed peptide scaffold when comparing the X-mean (Tables S8–11†). This gives an indication that the covalently attached peptide reduced the change to the activated myofibroblast phenotype and supported increased ALDH expression.
In general terms, these data were in accord with the improved results in 3D fibrous scaffolds for cell culture and tissue engineering compared to 2D cultures reported in recent years.39–43 The fibrous structure in these materials was designed to mimic ECM properties, resulting in an increase in desired cell response. However, as the synthetic polymers used in our culture assays were non-native to the cells, we anticipated that peptide or protein incorporation or functionalisation would be required to overcome some key practical challenges. These included poor cell attachment and induction of inflammation or undesired cell differentiation due to the sub-optimal mechanical or chemical properties (such as stiffness and hydrophobicity) of conventional polymer fibres.39–43 For example, Rodina et al.42 investigated migration and proliferation of mesenchymal stromal stem cells (MSSCs) on collagen protein modified PLA fibrous scaffolds. They observed more uniform distribution and cell penetration into these modified scaffolds. In addition to proteins, more specific peptide sequences can be used as ligands for enhanced cell attachment. The Arg-Gly-Asp (RGD) collagen/fibronectin peptide sequence is the most commonly used peptide incorporated in biomaterials to increase their cell adhesion properties.44 Besides, incorporation of known cell attachment peptide sequences (such as RGD), more cell specific peptide sequences can be incorporated to improve selective cell function or differentiation on the materials.45 Examples include the laminin-derived peptides IKVAV46 and YIGSR47 for cell attachment and anti-cancerous response, specific bone morphogenetic protein-2-related peptide P24 and rhBMP2,48,49 MMP degradable peptide sequences50 and enzyme responsive peptides (such as Fmoc-propargyl-GAARGD, which can be cleaved to ARGD by the enzyme elastase expressed by porcine pancreas).51
In this study, we set out a new approach for 3D mammalian cell culture by functionalising thermo-responsive fibrous scaffolds with a cell responsive peptide sequence. The methodology employed here to functionalise the scaffolds is amenable to the attachment of other peptide sequences as desired. In our case the laminin-1 peptide component, YIGSR, known to promote hCSCs interaction and to support the quiescent keratocyte culture on the scaffolds was chosen as an exemplar.22
Previous studies have demonstrated that laminin improves growth, organisation and differentiation of many cell types.22,52–55 Nonetheless, the use of laminin has many disadvantages such as degradation in culture to inactive fragments, tumorigenesis and induction of immune-responses, which make the full sequence unsuitable for clinical application.56 This risk can be minimised by selecting small peptide sequences within the laminin structure, which have the ability to improve adhesion and cell migration. One of these sequences is YIGSR, which has been observed to improve cell adhesion and migration.57–61 For example, Uzunalli et al.22 investigated the response of hCSCs on bioactive self-assembled peptide nanofibres. Two peptide sequences were investigated; (1) YIGSR (laminin derived) and (2) RGD (fibronectin derived). In both these cases, hCSCs were observed to maintain their characteristic morphology, and their proliferation was enhanced when YIGSR was present. Significantly, less cell proliferation and adhesion was observed for the RGD fibres. This study suggested that the YIGSR sequence has a direct impact on hCSCs attachment, proliferation and migration on 3D structures. These observations are in line with the increase in proliferation of the hCSCs observed on our scaffolds with increasing molar ratio of GGG-YIGSR covalently bound on the surface. Many studies have reported improved attachment and proliferation of human cornea epithelial cells.61–65 However, only one paper has described the response of hCSCs on YIGSR incorporated scaffolds,22 and no phenotypic support study has been performed to our knowledge. The immuno-staining and flow cytometry data reported here suggests that not only does YIGSR enhance cell attachment and proliferation on the fibrous scaffold, but the peptide also supports a higher percentage of the cell population which expresses a quiescent phenotype. Down-regulation of the undesired myofibroblastic marker α-SMA was detected, while up-regulation of keratocyte markers CD34 and ALDH was observed. Phenotypic characteristics were observed to remain constant when cultured and passaged, by the thermo-responsive enzymatic free passaging capabilities of the scaffolds. This gives an indication that such scaffolds can be designed to support the desired cell phenotype during in vitro culture and enzyme-free passaging and therefore this system is suitable for therapeutically relevant cell types.
Conclusions
The peptide functionalised, thermo-responsive electrospun scaffolds described here were designed for human cornea stromal cells (hCSCs) proliferation, phenotype support and enzymatic digestion-free passaging. The initially proposed scaffolds (PLA with 10 wt% PDEGMA) did not facilitate hCSCs adhesion and proliferation. Therefore, a peptide-functionalised scaffold was produced to improve the attachment of hCSCs on the scaffold and support the desired quiescent phenotype. PDEGMA/SH polymers were accordingly synthesised, their thermo-responsive properties and thiol content confirmed, and the materials were successfully blend-electrospun with PLA into fibrous scaffolds. The presence and homogenous distribution of free thiol on the scaffold surfaces, and the subsequent conjugation of Nor-GGG-YIGSR was confirmed by ToF-SIMS analysis. Non-specific attachment of the peptide was observed on thermo-responsive fibres without free thiol functionality but microscopy studies showed aggregated cells on these scaffolds, while elongated and spread cell morphologies were observed on thiol-containing and covalently-bound peptide derivatised scaffolds. These data indicated that enhanced hCSCs interaction was dependent on covalent peptide-functionalisation of the thermo-responsive fibres. The undesired, activated myofibroblastic phenotype was observed to decrease with increased molar percentage of Nor-GGG-YIGSR conjugated polymers at the scaffold's surface. The highest levels of peptide-functionalisation resulted in a decrease in keratocyte specific markers ALDH and CD34, whereas concentrations of 3 and 4 mol% Nor-GGG-YIGSR resulted in the highest expression of keratocyte markers, while suppressing the active myofibroblastic phenotype. Between 80–100% of cultured cells were observed to detach from the scaffolds at temperatures below the phase transition temperature of the thermo-responsive polymers at the surfaces of the fibres. Despite some variation in detachment of the cells from the scaffolds, cells were able to retain their cell phenotype after thermal cycles of attachment and detachment passaging. Hence, it can be concluded that the designed peptide conjugated thermo-responsive fibrous scaffolds are suitable for the attachment, proliferation, quiescent keratocyte phenotype support and temperature-mediated-passaging of hCSCs. In our studies, we used a peptide sequence to aid in hCSCs cell culture and expansion, but the attached peptide sequence could easily be changed to select for a different cell type, making the fabricated scaffolds a versatile matrix for cell expansion and phenotype support of any cell type including those destined for the clinic.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council [grant number EP/F500491/1] in the EPSRC Centre of Doctoral Training in Regenerative Medicine. The authors would like to thank Jiang Long and David Scurr for the help in ToF-SIMS and Emily Smith and Marta Alvarez Paino for conducting the XPS measurements and analysis, as well as, Hang Lau, Tianzhi Luo and Michael Haider for their advice and help during the work conducted at the University of Delaware in Prof. Kiick, Prof. Pochan and Dr Martin's labs.Notes and references
- K. Duval, H. Grover, L. H. Han, Y. Mou, A. F. Pegoraro, J. Fredberg and Z. Chen, Physiology, 2017, 32, 266–277 CrossRef CAS PubMed.
- R. D. Xu, M. B. Taskin, M. Rubert, D. Seliktar, F. Besenbacher and M. L. Chen, Sci. Rep., 2015, 5, 8480 CrossRef CAS PubMed.
- C. M. Madl, S. C. Heilshorn and H. M. Blau, Nature, 2018, 557, 335–342 CrossRef CAS PubMed.
- S. Vedicherla and C. T. Buckley, BioMed Res. Int., 2017, 2017, 2395138 Search PubMed.
- H. L. Huang, H. W. Hsing, T. C. Lai, Y. W. Chen, T. R. Lee, H. T. Chan, P. C. Lyu, C. L. Wu, Y. C. Lu, S. T. Lin, C. W. Lin, C. H. Lai, H. T. Chang, H. C. Chou and H. L. Chan, J. Biomed. Sci., 2010, 17, 36–46 CrossRef PubMed.
- P. Danhier, T. Copetti, G. De Preter, P. Leveque, O. Feron, B. F. Jordan, P. Sonveaux and B. Gallez, PLoS One, 2013, 8, e53324 CrossRef CAS PubMed.
- F. Gambinossi, L. S. Sefcik, E. Wischerhoff, A. Laschewsky and J. K. Ferri, ACS Appl. Mater. Interfaces, 2015, 7, 2518–2528 CrossRef CAS PubMed.
- S. Nara, S. Chameettachal, S. Midha, H. Singh, R. Tandon, S. Mohanty and S. Ghosh, J. Mater. Chem. B, 2015, 3, 4155–4169 RSC.
- K. Itoga and T. Okano, J. Mater. Chem., 2010, 20, 8768–8775 RSC.
- S. Dey, B. Kellam, M. R. Alexander, C. Alexander and F. R. A. J. Rose, J. Mater. Chem., 2011, 21, 6883–6890 RSC.
- X. Xue, L. Thiagarajan, S. Braim, B. R. Saunders, K. M. Shakesheff and C. Alexander, J. Mater. Chem. B, 2017, 5, 4926–4933 RSC.
- A. Saeed, N. Francini, L. White, J. Dixon, T. Gould, H. Rashidi, R. C. Al Ghanami, V. Hruschka, H. Redl, B. R. Saunders, C. Alexander and K. M. Shakesheff, Adv. Mater., 2015, 27, 662–668 CrossRef CAS PubMed.
- S. A. Braim, K. M. Shakesheff, B. R. Saunders and C. Alexander, J. Mater. Chem. B, 2016, 4, 962–972 RSC.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- N. Badi, Prog. Polym. Sci., 2017, 66, 54–79 CrossRef CAS.
- M. Ulasan, E. Yavuz, E. U. Bagriacik, Y. Cengeloglu and M. S. Yavuz, J. Biomed. Mater. Res., Part A, 2015, 103, 243–251 CrossRef PubMed.
- S. M. Hashemnejad, A. Z. M. Badruddoza, B. Zarket, C. Ricardo Castaneda and P. S. Doyle, Nat. Commun., 2019, 10, 2749 CrossRef PubMed.
- Y. Yao, Y.-Z. Ma, M. Qin, X.-J. Ma, C. Wang and X.-Z. Feng, Colloids Surf., B, 2008, 66, 233–239 CrossRef CAS PubMed.
- A. M. Aladdad, M. H. Amer, L. Sidney, A. Hopkinson, L. J. White, C. Alexander and F. R. A. J. Rose, Acta Biomater., 2019, 95, 427–438 CrossRef CAS.
- C. Zhong, J. Wu, C. A. Reinhart-King and C. C. Chu, Acta Biomater., 2010, 6, 3908–3918 CrossRef CAS PubMed.
- L. E. Sidney and A. Hopkinson, J. Tissue Eng. Regener. Med., 2018, 12, e203–e215 CrossRef CAS PubMed.
- G. Uzunalli, Z. Soran, T. S. Erkal, Y. S. Dagdas, E. Dinc, A. M. Hondur, K. Bilgihan, B. Aydin, M. O. Guler and A. B. Tekinay, Acta Biomater., 2014, 10, 1156–1166 CrossRef CAS PubMed.
- L. E. Sidney, M. J. Branch, H. S. Dua and A. Hopkinson, Cytotherapy, 2015, 17, 1706–1722 CrossRef CAS PubMed.
- K. Hashmani, M. J. Branch, L. E. Sidney, P. S. Dhillon, M. Verma, O. D. McIntosh, A. Hopkinson and H. S. Dua, Stem Cell Res. Ther., 2013, 4, 226–226 Search PubMed.
- F. A. A. Ruiter, C. Alexander, F. R. A. J. Rose and J. I. Segal, Biomed. Mater., 2017, 12, 055009 CrossRef CAS PubMed.
- P. Kingshott, S. McArthur, H. Thissen, D. G. Castner and H. J. Griesser, Biomaterials, 2002, 23, 4775–4785 CrossRef CAS PubMed.
- C. M. Mahoney, S. V. Roberson and G. Gillen, Anal. Chem., 2004, 76, 3199–3207 CrossRef CAS PubMed.
- D. Cossement, R. Gouttebaron, V. Cornet, P. Viville, M. Hecq and R. Lazzaroni, Appl. Surf. Sci., 2006, 252, 6636–6639 CrossRef CAS.
- H. K. Shon, M. Son, K. M. Park, C. K. Rhee, N. W. Song, H. M. Park, D. W. Moon and T. G. Lee, Surf. Interface Anal., 2011, 43, 628–631 CrossRef CAS.
- Y. Yokoyama, S. Aoyagi, M. Fujii, J. Matsuo, J. S. Fletcher, N. P. Lockyer, J. C. Vickerman, M. K. Passarelli, R. Havelund and M. P. Seah, Anal. Chem., 2016, 88, 3592–3597 CrossRef CAS PubMed.
- M. Körsgen, A. Pelster, K. Dreisewerd and H. F. Arlinghaus, J. Am. Soc. Mass Spectrom., 2016, 27, 277–284 CrossRef PubMed.
- S. Ying, A. Shiraishi, C. W. Kao, R. L. Converse, J. L. Funderburgh, J. Swiergiel, M. R. Roth, G. W. Conrad and W. W. Kao, J. Biol. Chem., 1997, 272, 30306–30313 CrossRef CAS PubMed.
- S. Saika, A. Shiraishi, C. Y. Liu, J. L. Funderburgh, C. W. C. Kao, R. L. Converse and W. W. Kao, J. Biol. Chem., 2000, 275, 2607–2612 CrossRef CAS PubMed.
- K. Musselmann, B. Alexandrou, B. Kane and J. R. Hassell, J. Biol. Chem., 2005, 280, 32634–32639 CrossRef CAS.
- T. Sumide, K. Nishida, M. Yamato, T. Ide, Y. Hayashida, K. Watanabe, J. Yang, C. Kohno, A. Kikuchi, N. Maeda, H. Watanabe, T. Okano and Y. Tano, FASEB J., 2006, 20, 392–394 CrossRef CAS.
- J. Teichmann, M. Nitschke, D. Pette, M. Valtink, S. Gramm, F. V. Hartel, T. Noll, R. H. W. Funk, K. Engelmann and C. Werner, Sci. Technol. Adv. Mater., 2015, 16, 045003–045003 CrossRef.
- Y.-F. Li, P. Slemming-Adamsen, J. Wang, J. Song, X. Wang, Y. Yu, M. Dong, C. Chen, F. Besenbacher and M. Chen, J. Tissue Eng. Regener. Med., 2017, 11, 2411–2420 CrossRef CAS.
- A. M. Aladdad, M. H. Amer, L. Sidney, A. Hopkinson, L. J. White, C. Alexander and F. R. A. J. Rose, Acta Biomater., 2019, 95, 427–438 CrossRef CAS PubMed.
- N. Masoumi, D. Copper, P. Chen, A. Cubberley, K. Guo, R. Z. Lin, B. Ahmed, D. Martin, E. Aikawa, J. Melero-Martin and J. Mayer, Adv. Funct. Mater., 2017, 27, 1606614–1606614 CrossRef.
- J. M. Holzwarth and P. X. Ma, J. Mater. Chem., 2011, 21, 10243–10251 RSC.
- T. D. Stocco, B. V. M. Rodrigues, F. R. Marciano and A. O. Lobo, Mater. Lett., 2017, 196, 221–224 CrossRef CAS.
- A. V. Rodina, T. K. Tenchurin, V. P. Saprykin, A. D. Shepelev, V. G. Mamagulashvili, T. E. Grigor'ev, K. I. Lukanina, A. S. Orekhov, E. Y. Moskaleva and S. N. Chvalun, Bull. Exp. Biol. Med., 2016, 162, 120–126 CrossRef CAS PubMed.
- J. M. Lee, T. Chae, F. A. Sheikh, H. W. Ju, B. M. Moon, H. J. Park, Y. R. Park and C. H. Park, Mater. Sci. Eng., C, 2016, 68, 758–767 CrossRef CAS PubMed.
- N. J. Hogrebe and K. J. Gooch, J. Biomed. Mater. Res., Part A, 2016, 104, 2356–2368 CrossRef CAS PubMed.
- N. Huettner, T. R. Dargaville and A. Forget, Trends Biotechnol., 2018, 36, 372–383 CrossRef CAS PubMed.
- A. Farrukh, S. F. Zhao, J. I. Paez, A. Kavyanifar, M. Salierno, A. Cavalie and A. del Campo, ACS Appl. Mater. Interfaces, 2018, 10, 41129–41137 CrossRef CAS PubMed.
- L. M. Nibourg, E. Gelens, S. A. F. Nibourg, M. R. de Jong, R. Kuijer, T. G. van Kooten and S. A. Koopmans, Acta Ophthalmol., 2016, 94, 721–729 CrossRef CAS PubMed.
- Z. H. Deng, Y. S. Li, X. Gao, G. H. Lei and J. Huard, Osteoarthritis Cartilage, 2018, 26, 1153–1161 CrossRef CAS PubMed.
- J. F. Li, L. Jin, M. B. Wang, S. B. Zhu and S. Y. Xu, Biomed. Mater., 2015, 10, 045004 CrossRef PubMed.
- J. Yu, F. Chen, X. Wang, N. Dong, C. Lu, G. Yang and Z. Chen, Polym. Degrad. Stab., 2016, 133, 312–320 CrossRef CAS.
- J. N. Roberts, J. K. Sahoo, L. E. McNamara, K. V. Burgess, J. L. Yang, E. V. Alakpa, H. J. Anderson, J. Hay, L. A. Turner, S. J. Yarwood, M. Zelzer, R. O. C. Oreffo, R. V. Ulijn and M. J. Dalby, ACS Nano, 2016, 10, 6667–6679 CrossRef CAS PubMed.
- A. M. Baur, T. I. Gamberger, H. G. Weerda, M. Gjuric and E. R. Tamm, Acta Otolaryngol., 1995, 115, 517–521 CrossRef CAS PubMed.
- K. Stamati, J. V. Priestley, V. Mudera and U. Cheema, Exp. Cell Res., 2014, 327, 68–77 CrossRef CAS PubMed.
- C. L. Maruyama, N. J. Leigh, J. W. Nelson, A. D. McCall, R. E. Mellas, P. Lei, S. T. Andreadis and O. J. Baker, J. Dent. Res., 2015, 94, 1610–1617 CrossRef CAS PubMed.
- C. V. Leiton, A. Aranmolate, C. Eyermann, M. J. Menezes, L. F. Escobar-Hoyos, S. Husain, S. J. Winder and H. Colognato, J. Neurochem., 2015, 135, 522–538 CrossRef CAS PubMed.
- P. Topley, D. C. Jenkins, E. A. Jessup and J. N. Stables, Br. J. Cancer, 1993, 67, 953–958 CrossRef CAS PubMed.
- Y. Hosokawa, Y. Takahashi, Y. Kadoya, S. N. M. Yamashina, Y. Yamada and H. Nogawa, Dev., Growth Differ., 1999, 41, 207–216 CrossRef CAS PubMed.
- Y. Yamada, K. Hozumi, F. Katagiri, Y. Kikkawa and M. Nomizu, Biomaterials, 2013, 34, 6539–6547 CrossRef CAS.
- K. Nam, C. S. Wang, C. L. M. Maruyama, P. Lei, S. T. Andreadis and O. J. Baker, J. Dent. Res., 2017, 96, 798–806 CrossRef CAS.
- C. Kilic, A. Girotti, J. C. Rodriguez-Cabello and V. Hasirci, Biomater. Sci., 2014, 2, 318–329 RSC.
- X. D. Duan, C. McLaughlin, M. Griffith and H. Sheardown, Biomaterials, 2007, 28, 78–88 CrossRef CAS PubMed.
- K. Merrett, C. M. Griffith, Y. Deslandes, G. Pleizier and H. Sheardown, J. Biomater. Sci., Polym. Ed., 2001, 12, 647–671 CrossRef CAS PubMed.
- L. Aucoin, C. M. Griffith, G. Pleizier, Y. Deslandes and H. Sheardown, J. Biomater. Sci., Polym. Ed., 2002, 13, 447–462 CrossRef CAS PubMed.
- F. Li, D. Carlsson, C. Lohmann, E. Suuronen, S. Vascotto, K. Kobuch, H. Sheardown, R. Munger, M. Nakamura and M. Griffith, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15346–15351 CrossRef CAS.
- X. Duan and H. Sheardown, J. Biomater. Sci., Polym. Ed., 2007, 18, 701–711 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01965k |
| ‡ FAA Ruiter is now working at Maastricht University in the MERLN institute. |
| This journal is © The Royal Society of Chemistry 2020 |