
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Scaffold channel size influences stem cell differentiation pathway in 3-D printed silica hybrid scaffolds for cartilage regeneration
Siwei
Li
a,
Francesca
Tallia
 a,
Ali A.
Mohammed
a,
Molly M.
Stevens
abc and
Julian R.
Jones
*a
a,
Ali A.
Mohammed
a,
Molly M.
Stevens
abc and
Julian R.
Jones
*a
aDepartment of Materials, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: julian.r.jones@imperial.ac.uk
bDepartment of Bioengineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
cInstitute of Biomedical Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
First published on 14th February 2020
Abstract
We report that 3-D printed scaffold channel size can direct bone marrow derived stem cell differentiation. Treatment of articular cartilage trauma injuries, such as microfracture surgery, have limited success because durability is limited as fibrocartilage forms. A scaffold-assisted approach, combining microfracture with biomaterials has potential if the scaffold can promote articular cartilage production and share load with cartilage. Here, we investigated human bone marrow derived stromal cell (hBMSC) differentiation in vitro in 3-D printed silica/poly(tetrahydrofuran)/poly(ε-caprolactone) hybrid scaffolds with specific channel sizes. Channel widths of ∼230 μm (210 ± 22 μm mean strut size, 42.4 ± 3.9% porosity) provoked hBMSC differentiation down a chondrogenic path, with collagen Type II matrix prevalent, indicative of hyaline cartilage. When pores were larger (∼500 μm, 229 ± 29 μm mean strut size, 63.8 ± 1.6% porosity) collagen Type I was dominant, indicating fibrocartilage. There was less matrix and voids in smaller channels (∼100 μm, 218 ± 28 μm mean strut size, 31.2 ± 2.9% porosity). Our findings suggest that a 200–250 μm pore channel width, in combination with the surface chemistry and stiffness of the scaffold, is optimal for cell–cell interactions to promote chondrogenic differentiation and enable the chondrocytes to maintain their phenotype.
1. Introduction
Native adult articular cartilage lacks innervation and vascularisation. Chondrocytes, the cells responsible for extracellular matrix (ECM) homeostasis, are relatively quiescent and low in number.1 Therefore, this highly specialised connective tissue has limited intrinsic self-repair capacity, especially if a defect is confined to cartilage. Microfracture surgery is a technique used on patients with sports injuries, wherein a small (up to 2 cm2) defect is cleaned and holes are made in the subchondral bone to liberate bone marrow that contains stem cells. The clot produces new cartilage but long-term durability is limited as fibrous cartilage of inferior biomechanical properties is produced.2 A one-step scaffold-assisted regenerative approach, combining the microfracture with biomaterials has potential if the scaffold can promote collagen Type II matrix production and share load with the host cartilage. Autologous chondrocyte implantation (ACI) and related procedures3 are expensive and lengthy and while clinical studies have not demonstrated improvement over microfracture, they have prompted the emergence of autologous matrix-induced chondrogenesis (AMIC).4–6AMIC combines the microfracture surgical technique with the use of a biomaterial scaffold. A current device used in AMIC procedures is Chondro-Gide® (Geistlich Biomaterials, Switzerland), a bi-layer structure of porcine derived Type I/III collagen. Fibrin glue is used to adhere the scaffold to the lesion following microfracture perforations in subchondral bone.7 Although a pattern of positive patient outcomes can be drawn from clinical studies, the long term success of such material remains debatable.8–10 Many studies included patients that required additional surgical procedures such as osteotomies, it is therefore difficult to determine the benefit of AMIC alone. In addition, some studies reported patient outcomes declined significantly as early as after 1.5 years post-operation.10–12 Low durability of the repaired cartilage is attributed to the new cartilage being fibrocartilage-like, which has inferior mechanical properties to articular hyaline cartilage.13,14
Gels, such as alginate, have also been used have been used in modified AMIC trials in animal studies,15 however, hydrogels provide limited initial support due to inferior mechanical properties and further modifications, e.g. incorporation of matrix derived molecules such as collagen Type I or II, are often required for endogenous stem cell recruitment.16 An ideal scaffold inserted into the microfracture procedure should retain the cells in situ, provide mechanical support, and serve as a guide for articular cartilage formation. In monolayer conditions, chondrocytes undergo dedifferentiation and subsequently cease the production of matrix proteins such as aggrecan and collagen Type II during proliferation.17,18 Preventing this phenotype change during microfracture is crucial to achieve regeneration of hyaline cartilage and provide long-term success. Ensuring chondrocytes do not experience monolayer-like culture conditions within a scaffold is one consideration. In vitro efforts have been devoted to mimic the natural conditions e.g. replicating the 3-D environment (e.g. scaffold porosity)19,20 and mechanical properties;21,22 or the presence of hypoxic conditions23,24 and growth factors.25,26
We have previously described the syntheses of sol–gel derived hybrid materials that show potential for cartilage regeneration.27–29 Unlike composites, the inorganic and organic co-networks in hybrid materials interpenetrate and have covalent links between them, providing tailorable degradation characteristics and synergistic mechanical properties. The inorganic silica network provides stiffness, and the organic components, such as gelatin28,29 or poly(tetrahydrofuran) (PTHF) and poly(ε-caprolactone) (PCL-diCOOH),27 provide ductility, mimicking the native articular cartilage. The sol–gel methodology allows the hybrid materials to be directly 3-D printed without the addition of binders, allowing accurate control over scaffold design, porosity and mechanical properties.27 Optimisation of manufacturing process, in terms of the printing and mechanical properties for scaffolds with channel size of ∼200 μm was performed in previous work.27 Target channel sizes of ∼100 and ∼500 μm were also tested. Pores smaller than 100 μm cannot be printed, due to print nozzle limitations and beyond 500 μm, the pore channels warp as struts bow.
Previous work showed that in vitro cultures of ATDC5 murine chondrogenic cells in 3-D printed silica-poly(tetrahydrofuran)/poly(ε-caprolactone), silica-PTHF/PCL, scaffolds with ∼200 μm channel size produced hyaline cartilaginous matrix formation, i.e. collagen Type II matrix, with no collagen Type I or Type X produced.27 Expression of Sox9 and aggrecan were also enhanced. Scaffolds made solely of PCL with similar pore architectures did not provoke collagen Type II production.
A limitation of the previous study was that the ATDC5 cells are predisposed to forming a collagen Type II matrix. Here, the more clinically relevant human bone marrow derived stromal cells (hBMSCs) are investigated. A number of studies on other materials have investigated the effect of pore size on chondrogenesis, however due to large variations in experimental setup such as cell types, materials and structural design,19,30,31 it is not possible to draw meaningful conclusions. The aim of current study was to investigate the effect of channel size of the promising silica-PTHF/PCL scaffolds on differentiation of hBMSCs in chondrogenic media in vitro. The hypothesis tested was that, for scaffolds fabricated using silica-PTHF/PCL, channel size of ∼200 μm would favour hyaline cartilage matrix production over fibrocartilage or hypertrophic matrix production. Silica-PTHF/PCL scaffolds with channels of three pore sizes were 3-D printed.
2. Experimental section
Materials
All chemicals were purchased from Sigma-Aldrich and VWR, UK, and all cell culture reagents were obtained from Invitrogen and Sigma-Aldrich, UK, unless specified otherwise.Hybrid synthesis
Hybrid sol–gel containing silica (SiO2) as the inorganic network and PTHF/PCL-diCOOH as the organic component were prepared as described previously.27 The composition had an inorganic/organic wt% ratio of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :75, as developed in previous work,27 termed Si80-CL. Hybrid synthesis consisted of a two-step procedure. First, TEMPO oxidation was applied to PCL diol (average Mn = 530 Da), producing a dicarboxylic acid (PCL-diCOOH), which was then used in sol–gel hybrid synthesis. An organic precursor solution of PCL-diCOOH (1 mol), (3-glycidyloxypropyl)trimethoxysilane (GPTMS, 2 mol) and boron trifluoride diethyletherate (BF3·OEt2, 0.5 mol) in THF was prepared and stirred at room temperature for 1.5 h, during which polymerisation of THF to PTHF occurred. At the same time, the silica precursor tetraethyl orthosilicate (TEOS, 80 wt% with regards to PCL-diCOOH mass), was hydrolysed in a stoichiometric volume of deionised water by hydrochloric acid (1 M HCl, at a ratio of 1/3% v/v with respect to water). When the two separate solutions were fully reacted, the TEOS hydrolysis solution was added dropwise to the organic precursor solution and stirred at room temperature for 30 minutes to form the hybrid sol. Stirring was continued without a lid to evaporate part of the residual THF and accelerate the gelation process. Any bubbles were removed via an ultrasound bath.
:75, as developed in previous work,27 termed Si80-CL. Hybrid synthesis consisted of a two-step procedure. First, TEMPO oxidation was applied to PCL diol (average Mn = 530 Da), producing a dicarboxylic acid (PCL-diCOOH), which was then used in sol–gel hybrid synthesis. An organic precursor solution of PCL-diCOOH (1 mol), (3-glycidyloxypropyl)trimethoxysilane (GPTMS, 2 mol) and boron trifluoride diethyletherate (BF3·OEt2, 0.5 mol) in THF was prepared and stirred at room temperature for 1.5 h, during which polymerisation of THF to PTHF occurred. At the same time, the silica precursor tetraethyl orthosilicate (TEOS, 80 wt% with regards to PCL-diCOOH mass), was hydrolysed in a stoichiometric volume of deionised water by hydrochloric acid (1 M HCl, at a ratio of 1/3% v/v with respect to water). When the two separate solutions were fully reacted, the TEOS hydrolysis solution was added dropwise to the organic precursor solution and stirred at room temperature for 30 minutes to form the hybrid sol. Stirring was continued without a lid to evaporate part of the residual THF and accelerate the gelation process. Any bubbles were removed via an ultrasound bath.
3-D printing of porous SiO2/PTHF/PCL scaffolds
To produce the hybrid ink for 3-D printing, gelation was continued until a suitable viscosity was reached and the ink was transferred into a 3 mL Luer-Lock plastic syringe and residual air was carefully removed. The syringe was either used immediately or stored in a freezer at −82 °C. If frozen, the ink loaded in the syringe was let to thaw at room temperature (10–15 minutes). To obtain porous scaffolds, the syringe was equipped with a tapered tip for 3-D printing (Nordson EFD, UK) and was then placed into a robocasting machine (“Robocaster”, 3d Inks LLC, USA), connected to a computer equipped with the software “Robocad” (3d Inks LLC, USA), which controlled the printing of porous scaffolds following a specific CAD file. The gelation process continued in the syringe, gradually increasing the ink viscosity until the optimum for 3-D extrusion printing was achieved. This then allowed ∼1 h printing window, during which 3-D porous scaffolds were printed following an orthogonal grid-like pattern so that each layer consists of a linear array of parallel struts and alternating layers are oriented at 90° to each other without any shift. Scaffolds of the same design but with different vertical channel size were printed by changing the strut spacing, which was set to 0.50 mm, 0.60 mm and 1.0 mm respectively. In all three cases the following printing parameters were applied: conical nozzle with internal diameter of 0.20 mm, speed of 10 mm s−1, z-spacing of 0.21 mm. Scaffolds with side dimensions ranging between 10.0–12.0 mm and height of 4.2 mm were printed. Following printing of silica-PTHF/PCL hybrid, wet scaffolds were placed in Nalgene polymethylpentene (PMP) containers at 40 °C for ageing (3 days, sealed) and drying (gradual loosening of the lid over 4–7 days). At the end of the drying step, the shrinkage inherent in the sol–gel process determined the final dimensions of the scaffolds, which were then cut down to 5.0 mm side dimensions, with the final thickness ∼2.5 mm after shrinkage. Scaffolds were immersed in deionised water for 10 s to remove reaction by-products.Evaluation of 3-D printed scaffold architecture
Scaffold 3-D structure was investigated using SEM (JEOL 6010 LV, secondary electron imaging at a 20 kV accelerating voltage, using a working distance between 13–17 mm). Images of the top surface and of vertical cross-sections, which were exposed by sectioning the scaffolds with a sharp blade, were taken. Samples were coated with a 10 nm layer of chromium or gold in order to make them conductive. SEM images of the top surface of the scaffolds were analysed with ImageJ software to evaluate vertical channel size (i.e. the size of the pores in the direction of cell seeding) and strut size on the x–y plane (n ≥ 35). The skeletal density (ρsk) of the hybrid was known from helium pycnometry measurements (Ultrapycnometer-1000, Quantachrome Corporation, n = 20).27ρsk was used to calculate the percentage porosity in the scaffold with the following equation:where ρsc is the scaffold density calculated geometrically (scaffold mass over volume, including pores) (n = 5).
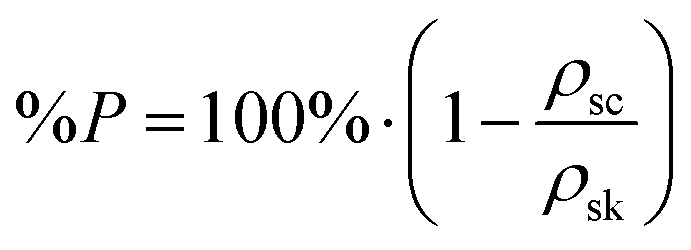
Chondrogenic differentiation on 3-D printed hybrid scaffolds
Commercially available hBMSCs were purchased (ATCC® PCS-500-012™, passage < 2) and monolayer expanded in basal conditions (α-MEM supplemented with 10% (v/v) foetal calf serum (FCS), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin) until confluence. Cells with passage number no more than 4 were used in the present study. Scaffolds (5 × 5 × 2.5 mm) with various pore sizes were sterilised with 70 vol% ethanol for 1 min and washed in phosphate buffered saline (PBS). hBMSCs were lysed from monolayer culture using trypsin-ethylenediaminetetraacetic acid (trypsin-EDTA) and suspended in basal media at a concentration of 1 × 106 cells in 5 mL. Cell suspension (5 mL) was added to each sterile 50 mL Falcon tube containing one scaffold. The tubes were placed in standard incubator for 2 h with gentle agitation every 30 minutes to allow diffused cell adhesion. The basal media was then replaced with chondrogenic media consisted of α-MEM supplemented with 10 ng mL−1 rhTGF-β3 (100-36E, PeproTech, UK), 100 μM L-ascorbic acid 2-phosphate, 10 nM dexamethasone and 1× ITS liquid supplement. Cultures were maintained for 21 days with medium change every 3–4 days.Immunohistochemistry
Following 21 days of culture, cell-scaffold constructs were collected and fixed in 4% paraformaldehyde (PFA). Samples were washed in PBS and cells permeabilised with buffered 0.5% Triton X-100 in buffered PBS (300 mM sucrose, 50 mM NaCl, 3 mM MgCl2, 20 mM HEPES and pH 7.2). Following blocking with 10 mg mL−1 bovine serum albumin (BSA) in PBS, samples were incubated with relevant primary antiserum and followed Alexa Fluor® 488-conjugated secondary antibody. Chondrogenic differentiation and cartilaginous matrix markers, anti-Sox9, Aggrecan, collagen Type II, collagen Type I and collagen Type X antibodies were used at dilutions of 1:150, 1:150, 1:500, 1:1000 and 1:100 respectively in 10 mg mL−1 BSA in PBS. All samples were counter-stained with DAPI (0.1 μg mL−1 in PBS).
Confocal microscopy
A Leica SP5 MP laser scanning confocal microscope (Leica Microsystems, Germany) was used for the imaging of central cross sections of stained scaffolds, cut using a sharp scalpel. Composite images were reconstructed using ImageJ software. The following settings for laser wavelengths were used: excitation at 488 nm/emission at 519 nm for Alexa Fluor® 488 and excitation at 405 nm/emission at 454 nm for DAPI–DNA complex.qPCR gene expression analysis
Cell-scaffold constructs were lysed for RNA extraction using Qiagen RNeasy kit (Qiagen, Manchester, UK) following manufacturer's instructions. Following treatment with DNase-1 reagent and RNA samples were reverse-transcribed using the SuperScript® VILO™ cDNA synthesis kit (Invitrogen, UK). SYBR green based qPCR assays were performed using the QuantStudio™ 6 Flex system (Thermo Fisher, UK). The following genes were analysed, Sox9 (F: 5′-cccttcaacctcccacacta-3′; R: 5′-tggtggtcggtgtagtcgta-3′), Aggrecan (F: 5′-gacggcttccaccagtgt-3′; R: 5′-gtctccatagcagccttcc-3′), Col2a1 (F: 5′-cctggtccccctggtcttgg-3′; R: 5′-catcaaatcctccagccatc-3′), Col1a1 (F: 5′-gagtgctgtcccgtctgc-3′; R: 5′-tttcttggtcggtgggtg-3′) and Col10a1 (F: 5′-cccactacccaacaccaaga-3′; R: 5′-gtggaccaggagtacc-3′). The expression of genes of interest was normalised to the housekeeping gene β-actin (F: 5′-ggcatcctcaccctgaagta-3′; R: 5′-aggtgtggtgccagattttc-3′). All primers were ordered from Sigma-Aldrich UK. The relative transcript levels of genes of interest were analysed using the comparative CT method (ΔΔCT method) according to “Applied Biosystems - Guide to Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR”. For each gene, the group with the highest expression was assigned a value of 1 and expression levels in the remaining groups were determined as fold relative to the group exhibiting the highest expression. Statistical analysis was performed at the level of ΔCT.Quantitative sulphated glycosaminoglycan (sGAG) assay
Total sGAG following 21-day culture on scaffolds was determined using the dimethylmethylene blue (DMMB) assay. Cell-seeded scaffolds were digested overnight at 60 °C in 0.7 U papain (in papain buffer consisting 8.2 mg mL−1 sodium acetate, 37 mg mL−1 disodium EDTA and 0.79 mg mL−1 cysteine hydrochloride in potassium phosphate solution containing 27.2 mg mL−1 monobasic potassium phosphate and 34.8 mg mL−1 dibasic potassium phosphate, pH adjusted to 6.4). The digested samples were centrifuged and the diluted extract (1:10 dilution in papain buffer) was then mixed with DMMB reagent (16 μg mL−1 DMMB, 2 mg mL−1 sodium formate, 0.5 vol% ethanol and 0.2 vol% formic acid) and optical density of the resultant solution was measured spectrophotometrically at 540 nm. The sGAG content of samples was extrapolated from a standard curve plotted from the optical density values of standard solutions of chondroitin sulphate from shark cartilage (concentration range: 0–100 μg mL−1).
Statistical analysis
Results were presented as mean ± S.D. Statistical analysis was performed using Mann–Whitney U test (2 groups) or Kruskal–Wallis test with Dunn's post test (3 or more groups) in Prism 7. Results were deemed significant if the probability of occurrence by random chance alone was less than 5% (i.e. p < 0.05).3. Results
3.1 Scaffold structure
An ink of silica-PTHF/PCL hybrid (with an inorganic:organic wt% ratio of 25:75) was printed in a regular grid-like pattern using 3-D extrusion printing (Robocasting)27 to produce scaffolds of three different channel widths (Fig. 1). Pore channels were visible from the top surface (Fig. 1a–c) and vertical sections (Fig. 1d–f), giving evidence of interconnection in all directions and confirming that that the extruded ink was able to hold its shape during printing. The scaffold struts had an irregular topography due to the extrusion process. Three vertical channel sizes were achieved (Table 1) by altering the strut spacing in the 3-D printing profile design file. Strut spacing is the distance between the central axes of adjacent parallel struts/filaments, which were set to 0.5 mm, 0.6 mm and 1.0 mm, resulting in final channel sizes of 117 ± 42 μm (SC-100), 231 ± 54 μm (SC-250) and 503 ± 82 μm (SC-500), respectively, due to the shrinkage caused by drying. The mean size of the struts ranged between 210–230 μm (Table 1) for all scaffolds, independent of the strut spacing as the nozzle diameter was the same for the three types of silica-PTHF/PCL scaffolds. Tallia et al. previously reported analysis of X-ray microcomputed tomography (μCT) images of the SC-250 scaffolds found channel sizes ranged from 40–240 μm.27 The mean channel and strut size reported here, measured from the top surface (i.e. x–y plane), are in the upper part of that range, and cells were seeded in this direction. Vertical cross-sections had channels and struts with lower height than width (Fig. 1). The percentage porosity (Table 1) increased from SC-100 to SC-500, as expected from increasing channel width (pore size) without modifying the strut diameter.
 | ||
| Fig. 1 SEM images of 3-D printed silica-PTHF/PCL hybrid scaffolds with different vertical channel widths: (a, d) images of SC-100 (mean channel width 117 ± 42 μm), printed with 0.5 mm strut spacing; (b, e) images of SC-250 (mean channel width 231 ± 54 μm), printed with 0.6 mm strut spacing; (c, f) images of SC-500 (mean channel width 503 ± 82 μm), printed with 1.0 mm strut spacing. a–b–c (top) shows images of the top surface; d–e–f (bottom) shows images of a vertical (z–y) section. Scale bars = 500 μm. | ||
| Strut spacing (mm) | Scaffold ID | Mean channel size (μm) | Mean strut size (μm) | Porosity (%) |
|---|---|---|---|---|
| 0.5 | SC-100 | 117 ± 42 | 218 ± 28 | 31.2 ± 2.9 |
| 0.6 | SC-250 | 231 ± 54 | 210 ± 22 | 42.4 ± 3.9 |
| 1.0 | SC-500 | 503 ± 82 | 229 ± 29 | 63.8 ± 1.6 |
3.2 hBMSCs on silica-PTHF/PCL hybrid scaffolds
Tallia et al. previously demonstrated that silica-PTHF/PCL scaffolds support cell attachment and differentiation of the ATDC5 murine chondrogenic cell line and, most importantly, production of hyaline-like cartilaginous matrix, when the pore channel size was ∼200 μm.27 Here, we evaluated chondrogenic differentiation of clinically relevant hBMSCs in 3-D printed silica-PTHF/PCL scaffolds, particularly the effect of scaffold pore channel size on hyaline cartilaginous matrix formation. hBMSCs with passage number of no more than 4 were used as expression of some stem cell markers, such as STRO-1, rapidly reduce after passage 6 and loss of expression was observed after passage 7.32 Chondrogenic differentiation and cartilaginous ECM formation were first assessed using immunohistochemistry (Fig. 2).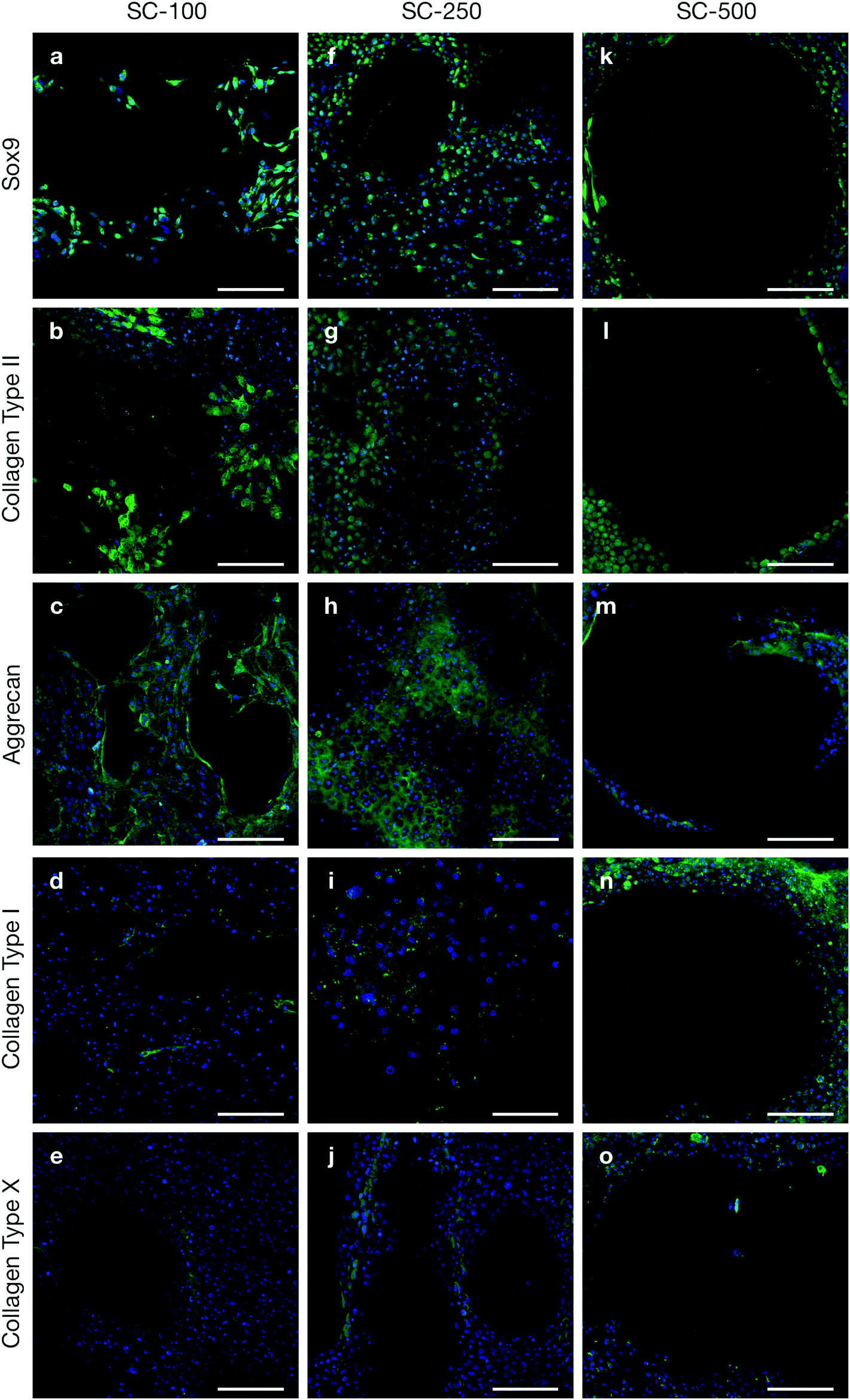 | ||
| Fig. 2 Immunohistochemical analysis of silica-PTHF/PCL hybrid scaffolds seeded with hBMSCs and cultured for 21 days in chondrogenic medium: (a–e) scaffolds with mean channel width of 117 ± 42 μm (SC-100); (f–j) scaffolds with mean channel width 231 ± 54 μm (SC-250); (k–o) scaffolds with mean channel width 503 ± 82 μm (SC-500). Cells cultured on both SC-100 and SC-250 hybrid scaffolds demonstrated chondrogenic differentiation, however hyaline cartilaginous ECM only distributed uniformly throughout SC-250 scaffolds. Sub-optimal chondrogenesis was observed on SC-500 scaffolds. Scale bar = 100 μm. | ||
hBMSCs cultured on both SC-100 and SC-250 scaffolds expressed Sox9, a marker for chondrogenic differentiation (Fig. 2a and f), plus markers related to hyaline cartilaginous ECM, including Collagen Type II (Fig. 2b and g) and aggrecan (Fig. 2c and h). In both scaffolds, positive collagen Type I staining was scarce (Fig. 2d and i). This indicates that the scaffolds provoked the cells to produce collagen Type II in preference to Type I. Within the SC-100 scaffolds, the pores appeared to be filled with collagen Type II matrix, however, there were also noticeable voids in the ECM, which were not seen for the SC-250 scaffolds. Sox9 and aggrecan were also distributed better throughout the pores in SC-250 compared to SC-100 scaffolds (Fig. 2a–c). Chondrogenic differentiation and ECM formation were suboptimal on SC-500 scaffolds (Fig. 2k–m). Although limited chondrogenic differentiation was observed, cells were preferentially distributed near the struts/walls of the SC-500 scaffolds, leaving most of the pore without positive staining for any of the markers tested, implying that ECM did not fill individual pores. While fibrous cartilage marker collagen Type I was present for all 3 channel widths of silica-PTHF/PCL scaffolds, its expression was particularly noticeable for hBMSCs cultured on SC-500 scaffolds, specifically along the struts of the scaffold structure (Fig. 2n). Expression of collagen Type X was observed in some cells on all pore size scaffolds (Fig. 2e, j and o).
qPCR analyses demonstrated that the expression of genes related to chondrogenic differentiation and hyaline cartilaginous ECM by hBMSCs on SC-100 and SC-250 scaffolds were significantly superior compared to those cultured on SC-500 scaffolds (Fig. 3). Cells cultured on SC-500 scaffolds expressed significantly less Sox9, Col2a1 and aggrecan and expressed significantly more Col1a1. hBMSCs cultured on SC-250 scaffolds did synthesise significantly more sGAG compared to those cultured on SC-500 scaffolds (Fig. 4). Although cells cultured on SC-250 scaffolds appeared to express more Sox9, Col2a1 and aggrecan than SC-100 scaffolds, the difference was not statistically significant. However, the amount of sGAG on SC-250 scaffolds also appeared to be higher compared to that on SC-100 scaffolds.
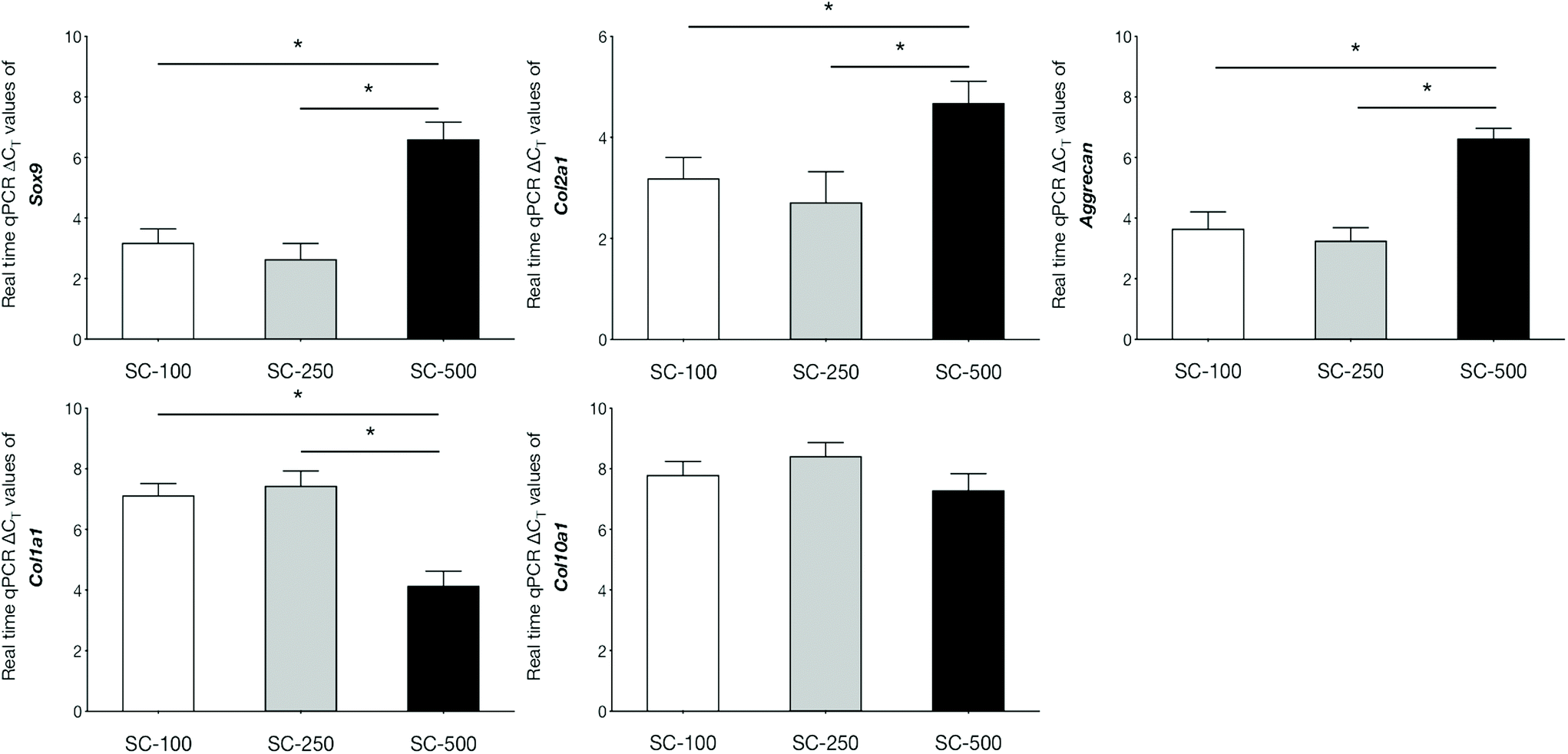 | ||
| Fig. 3 Analysis of gene expression by real-time qPCR at day-21 of hMSCs seeded in silica-PTHF/PCL hybrid scaffolds. For each gene, the group with the highest expression was assigned a value of 1 and expression levels in the remaining groups were determined using ΔΔCT method as fold relative to the group exhibiting the highest expression. Statistical analysis was performed at the level of ΔCT. * indicates P < 0.05. | ||
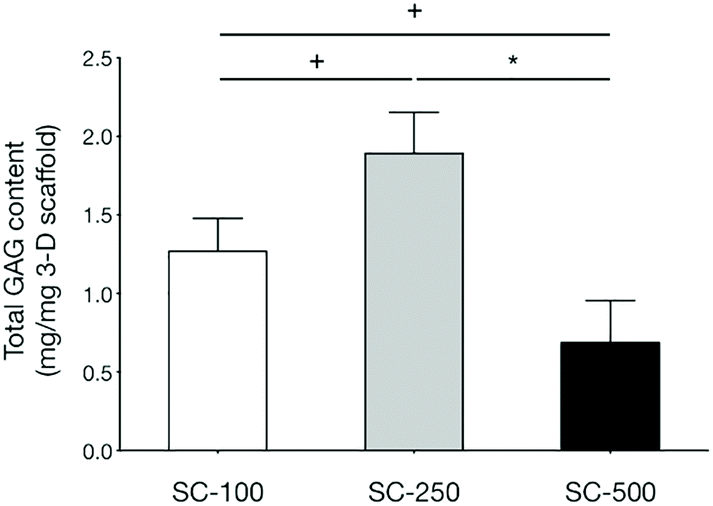 | ||
| Fig. 4 Analysis of sulphated GAG content in day-21 hBMSCs seeded in silica-PTHF/PCL hybrid scaffolds using the DMMB assay. Total sGAG content after 21 days of culture was normalised to the weight of scaffold at day 0. + indicates 0.05 < P < 0.1 and * indicates P < 0.05. | ||
4. Discussion
Scaffold architectural properties play important roles in cell distribution, mass transportation and formation of cartilaginous ECM.33,34 Although a number of studies have investigated the effect of pore size on chondrogenesis, due to large variations in cell types (e.g. bovine, human chondrocytes), materials (e.g. metal alloy, collagen) and structural design (e.g. foams, fibremats, sponges) investigated, it is not possible to draw meaningful conclusions from available literature. In a study that investigated the effect of pore size of a titanium alloy (Ti6Al4V) on ECM formation, with pore sizes of 13, 43 and 68 μm, the amount of sGAG accumulated per cell (bovine chondrocytes) was similar in all the constructs generated using scaffolds with different pore sizes.31 In a different study, significantly more collagen Type II was synthesised by bovine chondrocytes situated in the region with large pore size (1650 μm) compared to smaller pores within the same poly(ethylene glycol)-terephthalate–poly(butylene terephthalate) (PEGT/PBT) copolymer scaffolds that contained a gradient of pore sizes from 200 to 1650 μm.30 In another study, the amount of collagen Type II synthesised by immortalised human costal chondrocyte cell line was shown to be minimally affected by pore size and geometry of PCL scaffolds.19Here, hBMSCs were cultured on silica-PTHF/PCL scaffolds with target pore sizes in the seeding direction (i.e. z direction, x, y plane) of approximately 100, 250 and 500 μm. Our scaffolds, SC-100, SC-250 and SC-500 had mean vertical channel widths of ∼120, ∼230 and ∼500 μm (Fig. 1) respectively, maintaining strut diameter in the range 210–230 μm (Table 1) for all three types of scaffolds. The three types of scaffolds all had the same hybrid surface chemistry and inherent elastomeric mechanical properties.27 This allowed the current study to systemically evaluate solely the effect of pore size on chondrogenesis.
The results suggest hybrid scaffolds with pore sizes in the region of 200–250 μm achieved the balance between mass transportation and maintaining favourable 3-D environment for cell–cell interactions and subsequent chondrogenesis. Several studies, however, have reported formation of hyaline-cartilaginous matrix in scaffolds with pore sizes below 50 μm.31,35 Cell aggregation, such as those in a pellet culture model, is crucial to establish cell–cell interaction and an important cue for phenotypic expression of chondrocyte markers as evidenced in numerous previously published literature.35–37 It has also been suggested that reduced oxygen levels (hypoxia), due to increased diffusion barriers associated with smaller pore sizes, also favour chondrogenic differentiation and synthesis of cartilaginous ECM.20 However, the scaffolds used in these previous studies had significantly higher percentage porosity compared to 3-D printed silica-PTHF/PCL scaffolds used in the present study. Although chondrogenic differentiation and hyaline cartilaginous ECM formation was observed in silica-PTHF/PCL scaffolds with 100 μm pores (SC-100), ECM voids can be seen (Fig. 2) and quantitative qPCR and sGAG assay confirmed that chondrogenesis of hBMSCs were not as effective on SC-100 scaffolds in comparison to those in SC-250 scaffolds (Fig. 3 and 4). This was likely due to the less effective gas mass transport of gases and nutrients to cells and removal of catabolites from cells that was allowed by the SC-100 scaffolds used in the present study. It has been reported that substrate stiffness can control cellular behaviour and differentiation of stem cells without exogenous stimuli.38,39 Silica-PTHF/PCL scaffolds can support formation of hyaline cartilaginous matrix formation on scaffolds with appropriate pore size in the presence of chondrogenic supplements such as TGF-β3, further experiments and in vivo is required to determine whether the inherent stiffness of silica-PTHF/PCL hybrid has chondroinductive capabilities.
The progenitor/stem cells in the 500 μm pore channels may have differentiated into fibroblasts, or chondrocytes may have dedifferentiated to fibroblasts as they attached onto the pore walls and spread as if the walls were a 2-D surface. One possible approach to improve chondrogenesis in scaffolds with large 500 μm pores could be the use of significantly increased seeding cell number, as it is possible that increased seeding number can allow similar level of cell density achieved in smaller-sized pores with fewer chondrocytes.40 However, that high cell number may not be easily achieved in our proposed modality of cartilage repair, i.e. modified matrix assisted one-step microfracture procedure, as viable number of cells in the bone marrow are relatively low (1 cm3 of microfacture blood has ∼8000 CD34+ MSCs41) and may be further limited in patients with severely damaged articular cartilage or other underlying diseases. Low cell density can also cause loss of functional chondrocyte phenotype and in turn inadequate ECM synthesis.42 It was previously reported that spherical morphology of chondrocytes was maintained in high-density cultures, with spindle morphology observed in low-density cultures, but extremely high cell density could result in the formation of tight cell sheets or agglomerates, limiting oxygen, nutrient and waste product exchange, leading to apoptosis and reduced ECM formation.43,44 It has also been reported that in high cell density cultures, Sox9 could inhibit the expression of hyaline cartilage marker collagen Type II.45,46
In addition to cell–cell interaction, cell–material (cell–protein–material) interaction also plays an important role in maintaining phenotypic chondrocyte characteristics. Here, collagen Type I, a marker for fibrocartilage, was expressed by hBMSCs cultured in all silica-PTHF/PCL scaffolds, but was particularly noticeable in scaffolds with pore size of 500 μm (Fig. 2 and 3). It is likely that chondrocytes cultured on scaffolds with large pores considered the elongated struts a 2-D surface as shown in Scheme 1, and resumed elongated fibroblast-like morphology, a sign of dedifferentiation. In contrast, the restricted area of contact in smaller pore sized scaffolds may have facilitated the establishment of cell–cell contact amongst spherical shaped chondrocytes.
 | ||
| Scheme 1 Schematic drawing of the morphological changes of chondrocytes in scaffolds with different pore sizes: (a) scaffolds with ∼200 μm pores tend to facilitate cell–cell interaction and main spherical chondrocyte phenotype; (b) cells within larger spaces (e.g. 500 μm) flatten onto the struts of the scaffold as they would in a 2-D environment, assuming an elongated morphology, chondrocytes would potentially dedifferentiate. | ||
The reason for collagen Type I also being present in smaller pores could be that the hBMSCs contain a heterogeneous population of bone marrow stromal cells, many of which are lineage-committed progenitor cells in addition to a small proportion of stem cells.47,48 This could lead to some formation of fibrocartilage (collagen Type I) and hypertrophy (collagen Type X) even in conditions that are suitable for hyaline cartilage matrix production. Immunoselection from bone marrow mononuclear cells on the basis of positive expression of one or more established surface markers such as the STRO-1 antigen could be applied to isolate relatively homogenous populations of bone marrow-derived adult stem cells and potentially improve chondrogenesis.35,49 Such approach will be adopted in future studies and may accelerate the use of hybrid scaffolds in cartilage regeneration applications.
5. Conclusion
hBMSCs seeded on 3-D printed silica-PTHF/PCL hybrid scaffolds with pore channel width of approximately 200–250 μm preferentially supported chondrogenic differentiation from hBMSCs and hyaline cartilaginous ECM formation in vitro. Larger pores caused poor cell–cell interaction and dedifferentiation and smaller pores may not have had suitable space for matrix production.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the European Commission funding under the 7th Framework Programme (Marie Curie Innovative Training Network (ITN); grant number: 289958, Bioceramics for Bone Repair) and EPSRC (EP/M019950/1, EP/N025059/1). M. M. S. was funded by the grant from the UK Regenerative Medicine Platform “Acellular/Smart Materials – 3D Architecture” (MR/R015651/1). Raw data can be obtained from rdm-enquiries@imperial.ac.uk.References
- M. B. Goldring, Ther. Adv. Musculoskeletal Dis., 2012, 4, 269–285 CrossRef CAS PubMed.
- K. Mithoefer, T. McAdams, R. J. Williams, P. C. Kreuz and B. R. Mandelbaum, Am. J. Sports Med., 2009, 37, 2053–2063 CrossRef PubMed.
- S. Giannini, R. Buda, M. Cavallo, A. Ruffilli, A. Cenacchi, C. Cavallo and F. Vannini, Injury, 2010, 41, 1196–1203 CrossRef PubMed.
- S. Anders, M. Volz, H. Frick and J. Gellissen, Open Orthop. J., 2013, 7, 133–143 CrossRef PubMed.
- J. Chahla, R. F. LaPrade, R. Mardones, J. Huard, M. J. Philippon, S. Nho, O. Mei-Dan and C. Pascual-Garrido, Orthopedics, 2016, 39, e715–e723 CrossRef PubMed.
- U. Freymann, W. Petersen and C. Kaps, OA Orthopaedics, 2013, 1, 6 CrossRef.
- J. P. Benthien and P. Behrens, Knee Surg. Sports Traumatol. Arthrosc., 2011, 19, 1316–1319 CrossRef PubMed.
- N. Shaikh, M. K. T. Seah and W. S. Khan, World J Orthop., 2017, 8, 588–601 CrossRef PubMed.
- J. Schagemann, P. Behrens, A. Paech, H. Riepenhof, B. Kienast, H. Mittelstadt and J. Gille, Arch. Orthop. Trauma Surg., 2018, 138, 819–825 CrossRef CAS PubMed.
- J. Gille, E. Schuseil, J. Wimmer, J. Gellissen, A. P. Schulz and P. Behrens, Knee Surg. Sports Traumatol. Arthrosc., 2010, 18, 1456–1464 CrossRef CAS PubMed.
- R. Gudas, A. Gudaite, A. Pocius, A. Gudiene, E. Cekanauskas, E. Monastyreckiene and A. Basevicius, Am. J. Sports Med., 2012, 40, 2499–2508 CrossRef PubMed.
- R. Gudas, E. Stankevicius, E. Monastyreckiene, D. Pranys and R. J. Kalesinskas, Knee Surg. Sports Traumatol. Arthrosc., 2006, 14, 834–842 CrossRef PubMed.
- D. Mancini and A. Fontana, Int. Orthop., 2014, 38, 2057–2064 CrossRef PubMed.
- B. He, J. P. Wu, J. Xu, R. E. Day and T. B. Kirk, PLoS One, 2013, 8, e74303 CrossRef CAS PubMed.
- R. Baba, T. Onodera, M. Matsuoka, K. Hontani, Z. Joutoku, S. Matsubara, K. Homan and N. Iwasaki, Am. J. Sports Med., 2018, 46, 1970–1979 CrossRef.
- H. V. Almeida, B. N. Sathy, I. Dudurych, C. T. Buckley, F. J. O'Brien and D. J. Kelly, Tissue Eng., Part A, 2017, 23, 55–68 CrossRef CAS PubMed.
- M. Demoor, D. Ollitrault, T. Gomez-Leduc, M. Bouyoucef, M. Hervieu, H. Fabre, J. Lafont, J. M. Denoix, F. Audigie, F. Mallein-Gerin, F. Legendre and P. Galera, Biochim. Biophys. Acta, 2014, 1840, 2414–2440 CrossRef CAS PubMed.
- F. Zhang, Y. Yao, K. Su, P. X. Pang, R. Zhou, Y. Wang and D. A. Wang, Ann. Biomed. Eng., 2011, 39, 3042–3054 CrossRef PubMed.
- S. H. Oh, I. K. Park, J. M. Kim and J. H. Lee, Biomaterials, 2007, 28, 1664–1671 CrossRef CAS PubMed.
- M. M. Nava, L. Draghi, C. Giordano and R. Pietrabissa, J. Appl. Biomater. Funct. Mater., 2016, 14, e223–e229 CAS.
- C. G. Jeong and S. J. Hollister, Tissue Eng., Part A, 2010, 16, 3759–3768 CrossRef CAS PubMed.
- N. Isobe, T. Komamiya, S. Kimura, U. J. Kim and M. Wada, Int. J. Biol. Macromol., 2018, 117, 625–631 CrossRef CAS PubMed.
- D. S. Kim, Y. J. Ko, M. W. Lee, H. J. Park, Y. J. Park, D. I. Kim, K. W. Sung, H. H. Koo and K. H. Yoo, Cell Stress Chaperones, 2016, 21, 1089–1099 CrossRef CAS PubMed.
- S. Li, R. O. Oreffo, B. G. Sengers and R. S. Tare, Biotechnol. Bioeng., 2014, 111, 1876–1885 CrossRef CAS PubMed.
- G. Coricor and R. Serra, Sci. Rep., 2016, 6, 38616 CrossRef CAS PubMed.
- J. Fischer, M. Ortel, S. Hagmann, A. Hoeflich and W. Richter, J. Cell Physiol., 2016, 231, 2673–2681 CrossRef CAS PubMed.
- F. Tallia, L. Russo, S. Li, A. L. H. Orrin, X. Shi, S. Chen, J. A. M. Steele, S. Meille, J. Chevalier, P. D. Lee, M. M. Stevens, L. Cipolla and J. R. Jones, Mater. Horiz., 2018, 5, 849 RSC.
- O. Mahony, S. Yue, C. Ionescu, J. V. Hanna, M. E. Smith, P. D. Lee and J. R. Jones, J. Sol-Gel Sci. Technol., 2014, 69, 288–298 CrossRef CAS.
- O. Mahony, O. Tsigkou, C. Ionescu, C. Minelli, L. Ling, R. Hanly, M. E. Smith, M. M. Stevens and J. R. Jones, Adv. Funct. Mater., 2010, 20, 3835–3845 CrossRef CAS.
- T. B. Woodfield, C. A. Van Blitterswijk, J. De Wijn, T. J. Sims, A. P. Hollander and J. Riesle, Tissue Eng., 2005, 11, 1297–1311 CrossRef CAS PubMed.
- T. Bhardwaj, R. M. Pilliar, M. D. Grynpas and R. A. Kandel, J. Biomed. Mater. Res., 2001, 57, 190–199 CrossRef CAS.
- D. Gothard, K. Cheung, J. M. Kanczler, D. I. Wilson and R. O. Oreffo, Stem Cell Res. Ther., 2015, 6, 251 CrossRef.
- I. V. Ponomarev, L. M. Kochneva and D. Barnewitz, Bull. Exp. Biol. Med., 2014, 156, 548–555 CrossRef CAS PubMed.
- S. Nuernberger, N. Cyran, C. Albrecht, H. Redl, V. Vecsei and S. Marlovits, Biomaterials, 2011, 32, 1032–1040 CrossRef CAS PubMed.
- S. Li, B. G. Sengers, R. O. Oreffo and R. S. Tare, J. Biomater. Appl., 2015, 29, 824–836 CrossRef CAS PubMed.
- R. S. Tare, D. Howard, J. C. Pound, H. I. Roach and R. O. Oreffo, Biochem. Biophys. Res. Commun., 2005, 333, 609–621 CrossRef CAS PubMed.
- P. J. Emans, E. J. Jansen, D. van Iersel, T. J. Welting, T. B. Woodfield, S. K. Bulstra, J. Riesle, L. W. van Rhijn and R. Kuijer, J. Tissue Eng. Regener. Med., 2013, 7, 751–756 CrossRef CAS.
- R. Olivares-Navarrete, E. M. Lee, K. Smith, S. L. Hyzy, M. Doroudi, J. K. Williams, K. Gall, B. D. Boyan and Z. Schwartz, PLoS One, 2017, 12, e0170312 CrossRef PubMed.
- H. Lv, L. Li, M. Sun, Y. Zhang, L. Chen, Y. Rong and Y. Li, Stem Cell Res. Ther., 2015, 6, 103–114 CrossRef PubMed.
- A. Woods, G. Wang and F. Beier, J. Cell Physiol., 2007, 213, 1–8 CrossRef CAS PubMed.
- T. Tallheden, J. E. Dennis, D. P. Lennon, E. Sjogren-Jansson, A. I. Caplan and A. Lindahl, J. Bone Jt. Surg., Am. Vol., 2003, 85(Suppl 2), 93–100 CrossRef PubMed.
- D. R. Albrecht, G. H. Underhill, T. B. Wassermann, R. L. Sah and S. N. Bhatia, Nat. Methods, 2006, 3, 369–375 CrossRef CAS PubMed.
- S. Kobayashi, A. Meir and J. Urban, J. Orthop. Res., 2008, 26, 493–503 CrossRef CAS PubMed.
- K. Zhang, L. Wang, Q. Han, B. C. Heng, Z. Yang and Z. Ge, Biomed. Eng.: Appl. Basis Commun., 2013, 25, 1340001 Search PubMed.
- P. Bernstein, M. Dong, S. Graupher, D. Corbeil, M. Gelinsky, K.-P. Günther and S. Fickert, J. Biomed. Mater. Res., Part A, 2009, 91, 910–918 CrossRef PubMed.
- D. M. Bell, K. H. Leung, S. C. Wheatley, L. J. Ng, S. Zhou, K. W. Ling, M. H. Sham, P. Koopman, P. L. Tam and K. S. E. Cheah, Nat. Genet., 1997, 16, 174–178 CrossRef CAS PubMed.
- S. Post, B. M. Abdallah, J. F. Bentzon and M. Kassem, Bone, 2008, 43, 32–39 CrossRef CAS.
- J. I. Dawson and R. O. Oreffo, Arch. Biochem. Biophys., 2008, 473, 124–131 CrossRef CAS PubMed.
- R. S. Tare, P. D. Mitchell, J. Kanczler and R. O. Oreffo, Methods Mol. Biol., 2012, 816, 83–99 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |