
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional coatings combining bioactive peptides and affinity-based cytokine delivery for enhanced integration of degradable vascular grafts†
Franziska
Clauder
a,
Franziska D.
Zitzmann
b,
Sabrina
Friebe
 c,
Stefan G.
Mayr
c,
Andrea A.
Robitzki
b and
Annette G.
Beck-Sickinger
*a
c,
Stefan G.
Mayr
c,
Andrea A.
Robitzki
b and
Annette G.
Beck-Sickinger
*a
aInstitute of Biochemistry, Faculty of Life Sciences, Leipzig University, Brüderstrasse 34, 04103 Leipzig, Germany. E-mail: abeck-sickinger@uni-leipzig.de
bCentre for Biotechnology and Biomedicine (BBZ)/Institute of Biochemistry, Faculty of Life Sciences, Leipzig University, Deutscher Platz 5, 04103 Leipzig, Germany
cLeibniz-Institute of Surface Engineering (IOM), Permoserstraße 15, 04318 Leipzig, Germany
First published on 21st January 2020
Abstract
Insufficient endothelialization of cardiovascular devices is a high-risk factor for implant failure. Presentation of extracellular matrix (ECM)-derived coatings is a well-known strategy to improve implant integration. However, the complexity of the system is challenging and strategies for applying multifunctionality are required. Here, we engineered mussel-derived surface-binding peptides equipped with integrin (c[RGDfK]) and proteoglycan binding sites (FHRRIKA) for enhanced endothelialization. Surface-binding properties of the platform containing L-3,4-dihydroxyphenylalanine (DOPA) residues were confirmed for hydrophilized polycaprolactone-co-lactide scaffolds as well as for glass and polystyrene. Further, heparin and the heparin-binding angiogenic factors VEGF, FGF-2 and CXCL12 were immobilized onto the peptide in a modular assembly. Presentation of bioactive peptides greatly enhanced human umbilical vein endothelial cell (HUVEC) adhesion and survival under static and fluidic conditions. In subsequent investigations, peptide-heparin-complexes loaded with CXCL12 or VEGF had an additional increasing effect on cell viability, differentiation and migration. Finally, hemocompatibility of the coatings was ensured. This study demonstrates that coatings combining adhesion peptides, glycosaminoglycans and modulators are a versatile tool to convey ECM-inspired multifunctionality to biomaterials and efficiently promote their integration.
1. Introduction
According to the World Health Organization, diseases of the cardiovascular system are the number one cause of death worldwide. In 2016, they accounted for 31% of all global deaths and claimed 17.9 million lives.1 Cardiovascular diseases (CVDs) are a group of disorders of the heart and vasculature, treatments include dietary and lifestyle modification in the first place as well as pharmaceutical intervention. However, when this therapy fails, surgical intervention becomes necessary. For some CVDs, this requires medical devices such as prosthetic valves, patches or stents. Fabricating such devices from biodegradable polymers bears a number of advantages, the first and foremost being a “traceless” tissue regeneration. This is especially beneficial for children, where multiple interventions are often required during adolescence. Complete resorption of the scaffold allows an unobstructed integrity of the vessel wall, recovery of the vasomotor function and the absence of artifacts in non-invasive imaging techniques.2 By virtue of the numerous benefits in comparison to conventional materials, extensive research has been going into the development of such devices leading to the first FDA-approved biodegradable coronary artery stent in 20163 and indicating many more to come. Polycaprolactone (PCL) and its copolymers are among the most extensively used resorbable implant materials. It is less brittle than other polyesters and displays superior rheological and viscoelastic properties.4,5 In combination with modern fabrication techniques, an endless number of shapes can be produced enabling customized implant creation.6 However, challenges include the maintenance of strength and stability during wound healing and the long degradation time of 2–4 years. The latter can be fine-tuned by copolymerization with lactones or glycolides and lactides, which significantly shorten the resorption duration to suit the respective tissue regeneration times.4,5Other challenges for the application of PCL in tissue regeneration include its unfavorable surface properties for cell adhesion. The material is very hydrophobic and has a low wettability limiting cell–surface-interactions. An incomplete endothelial lining can cause platelet activation and clotting, leading to thrombosis, or the activation of smooth muscle cell proliferation, inducing intimal hyperplasia, two of the main reasons for implant failure.7 Therefore, the formation of new tissue is crucial to implant success and should be actively promoted by the graft. A promising approach to guide the regeneration process is inspired by the extracellular matrix (ECM). Herein, the structural components express a number of bioactive domains, that either bind to cell surface receptors, to other structural proteins or to signaling molecules, serving not only as a scaffold, but also as a mediator of cell activation status.8 Imitating this complex network, in a defined and controllable manner, on the implant surface is therefore of high interest. Possible alternatives to direct the incorporation of functional groups during the fabrication of the scaffold are surface-binding peptide coatings. Mussel-derived peptides containing L-3,4-dihydroxyphenylalanine residues, originally found in the byssus of Mytilus edulis,9 can mediate peptide binding to various materials.10 Functionalization with short adhesion peptides can then specifically guide cell attachment to the graft.11 Attracting the desired cell type for implant integration and discriminating over unspecific adsorption can be achieved with integrin-specific RGD peptides. The cyclic peptide c[RGDfK] has a high affinity towards integrin αVβ3, which is highly expressed on endothelial cells,12 but not towards integrin αIIbβ3, presented by platelets.13 The heparin-binding peptide FHRRIKA, derived from bone sialoprotein,14 also mediates cell adhesion by cell surface proteoglycans, but can likewise be used to immobilize soluble heparin. Owing to its excellent anticoagulant properties,15 heparin grafting itself could reduce thrombogenicity of the scaffold16 but further exploitation as a delivery system for heparin-binding cytokines could additionally modulate the healing response.17 Heparin and heparan sulfate bind almost all angiogenic cytokines with high affinity, stabilizing and sequestering them to their respective receptors.18 Among them, FGF-2 and VEGF represent key players stimulating migration, proliferation and survival of endothelial cells.19 The chemokine CXCL12, another pro-angiogenic factor, guides the recruitment of hematopoietic stem and progenitor cells,20–22 providing a potential source of endothelial progenitor cells (EPC) for tissue vascularization.23
Our objective was to determine whether coatings combining short adhesion peptides, GAGs and cytokines will effectively enhance cell–surface-interactions for biodegradable scaffolds. In the presented study, we examined a mussel-inspired surface binding approach for the generation of multifunctional peptide-based coatings. Bioactive peptides were synthesized combining solid-phase peptide synthesis and orthogonal click reactions. The coating affinity to polycaprolactone-co-lactide (PCLLC) scaffolds was verified. Further, the proteoglycan-binding properties of the coatings were exploited for the immobilization of heparin and heparin-binding cytokines. Finally, improvements in endothelialization of coated PCLLC were examined and general hemocompatibility ensured. The here identified principles, comprising the versatility of the peptide binding and the modularity of the components, can be applied to other materials and tissues expanding the scope of the coating.
2. Experimental procedures
2.1 Peptide synthesis
Surface-coating peptides were synthesized as described by Pagel et al.11 Briefly, the peptide MP was elongated on a TentaGel S RAM resin (IRIS Biotech) using standard Fmoc/tBu conditions. Amino acids were activated with equimolar amounts of HOBt and DIC, Fmoc-NH-(PEG)2-COOH (13 atoms) was activated with HATU and DIPEA. Following selective side-chain deprotection of Lys(Dde), the diene 5-[4-(1,2,4,5-tetrazin-3-yl)benzyl-amino]-5-oxopentanoic acid (Sigma-Aldrich) was introduced. The cyclic RGD peptide was synthesized according to Hassert et al.24 with the following adjustments: peptide elongation was carried out on 2-chlorotrityl chloride resin (Merck), cleaved off fully-protected using glacial acetic acid, TFE and DCM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:8, v/v) and cyclized in DCM using HOBt and DIC. The heparin-binding peptides were produced by automated solid-phase peptide synthesis (Syro peptide synthesizer, MultiSynTech) on Rink amide resin (IRIS Biotech). N-terminal functionalization with Fmoc-L-Lys(N3)-OH was carried out manually. For conjugation of the cyclic RGD peptide by Diels–Alder reaction with inverse electron demand (DARinv), TentaGel Resin loaded with diene-modified MP was swollen in water. Dienophile-functionalized RGD peptide was subsequently added and the reaction carried out for at least 5 h. The Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) was likewise performed on resin. MP or MP-RGD were swollen in water. After pre-incubation of CuSO4, TCEP and THPTA, HBP(2)-N3 was added and the pH adjusted to 8. After degassing with Ar, the reaction mixture was added to the resin and incubated for 24 h at 45 °C, shaking. Following filtration, the resin was washed and incubated with EDTA to remove excess copper. The biotinylated peptides Bio-MP(+) and Bio-MP(−) were synthesized on Rink amide resin analogous to MP, N-terminally elongated twice with Fmoc-L-Ahx-OH and finally biotinylated by HOBt/DIC activation in NMP.
:1:8, v/v) and cyclized in DCM using HOBt and DIC. The heparin-binding peptides were produced by automated solid-phase peptide synthesis (Syro peptide synthesizer, MultiSynTech) on Rink amide resin (IRIS Biotech). N-terminal functionalization with Fmoc-L-Lys(N3)-OH was carried out manually. For conjugation of the cyclic RGD peptide by Diels–Alder reaction with inverse electron demand (DARinv), TentaGel Resin loaded with diene-modified MP was swollen in water. Dienophile-functionalized RGD peptide was subsequently added and the reaction carried out for at least 5 h. The Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) was likewise performed on resin. MP or MP-RGD were swollen in water. After pre-incubation of CuSO4, TCEP and THPTA, HBP(2)-N3 was added and the pH adjusted to 8. After degassing with Ar, the reaction mixture was added to the resin and incubated for 24 h at 45 °C, shaking. Following filtration, the resin was washed and incubated with EDTA to remove excess copper. The biotinylated peptides Bio-MP(+) and Bio-MP(−) were synthesized on Rink amide resin analogous to MP, N-terminally elongated twice with Fmoc-L-Ahx-OH and finally biotinylated by HOBt/DIC activation in NMP.
The final cleavage and complete deprotection of the peptides was accomplished by incubation with TFA and scavenger (9:1, v/v) for 3 hours shaking at RT. The following scavenger mixtures were used: TA/TK (1:1, v/v) for c[RGDfK(dienophile)], H2O for HBP(2)-N3 and TA/EDT (7:3, v/v) for all mussel-derived peptides. Peptide purification was carried out on a Phenomenex Aeris Peptide XB-C18 column (100 Å, 5 μm, 250 × 21.2 mm) using linear gradients of Eluent B in Eluent A (A: 0.1% TFA in H2O, B: 0.08% TFA in ACN). Analytical identification of the peptides was performed by MALDI-ToF-MS (Bruker Daltonics) and ESI-Ion Trap MS (Bruker Daltonics) and observed m/z values matched the calculated molecular weights. Final purity ≥95% was confirmed by analytical HPLC on a Phenomenex Jupiter Proteo column (90 Å, 4 μm) and a Phenomenex Jupiter Proteo C18 (300 Å, 4 μm), using linear gradients of Eluent B in Eluent A.
2.2 Surface binding
Embroidered polycaprolactone-co-lactide (PCLLC) scaffolds, fabricated as described elsewhere,25 were kindly provided by Prof. Dr. Stefan Rammelt (TU Dresden, Germany) and treated with 1 M NaOH/50% MeOH (1:1, v/v) for 15 min for surface hydrophilization (PCLLCNaOH). To investigate the binding affinities of the different peptide derivatives, surfaces were incubated with dilutions of biotinylated peptides in 10 mM Tris buffer, pH 7.4. Subsequently, scaffolds were washed 4 times with TBS-T buffer (50 mM Tris, 150 mM NaCl, 0.1% Tween20, pH 7.6) and transferred to a new well. Detection of bound peptide was established via the biotin-tag in an ELISA-like assay, as described previously.24 In short, scaffolds were blocked with 10% BSA in TBS buffer (50 mM Tris, 150 mM NaCl, pH 7.6) following incubation with horseradish peroxidase-conjugated streptavidin (1:2000 in TBS containing 1% BSA). Detection was carried out using 3,3′,5,5′-tetramethylbenzidine (TMB), stopped with 1 M HCl and quantified by reading the absorption at 450 nm (Infinite M200, Tecan). Blank values derived from uncoated samples and were subtracted from the obtained data. The result is shown as mean + sem and represents three independent experiments performed in triplicates.
2.3 Heparin and cytokine binding to coated scaffolds
PCLLCNaOH scaffolds were coated with 1 μM peptide in 10 mM Tris buffer pH 7.4 overnight. Subsequently, wells were incubated with 1 μM biotinylated porcine heparin (bio-Hep, Sigma-Aldrich) in TBS for 1 h. Detection of bound bio-Hep was established via the biotin-tag as described previously.For cytokine immobilization, PCLLCNaOH scaffolds were coated with 1 μM peptide overnight following 1 μM heparin (Sigma-Aldrich) for 1 h. Next, scaffolds were blocked with 10% BSA in TBS and incubated with 1 μM CXCL12 or 0.1 μM FGF-2 and VEGF in TBS, respectively. Detection followed as described earlier by Pagel et al.11 using the respective primary antibodies (mouse anti-SDF-1, Santa Cruz Biotechnology, Inc.; mouse anti-human FGF-basic and mouse anti-human VEGF165, Peprotech) and a horseradish peroxidase-coupled anti-mouse binding protein (m-IgGκ BP-HRP, Santa Cruz Biotechnology, Inc.). The experiment was performed at least three times in triplicates and is presented as mean + sem.
2.4 Chemokine release
Coating with peptide, heparin and CXCL12 was performed as described above. Scaffolds were placed in reaction tubes filled with 300 μl PBS and incubated at 37 °C and shaking for the respective time points. After 3, 6, 9, 24, 48 and 72 h, the supernatant was collected and replaced with fresh PBS. The amount of CXCL12 present in each fraction was determined using the quantification kit “Human CXCL12/SDF-1 DuoSet ELISA” in combination with the “DuoSet Ancillary Reagent Kit” (both R&D Systems) according to the manufacturer's protocol. The data is represented as cumulative amounts released over time (mean ± sem) from three independent experiments performed in triplicates.2.5 Coating PCLLC scaffolds for cellular assays
PCLLCNaOH scaffolds were coated with 1 μM peptide in 10 mM Tris buffer pH 7.4 overnight. Unbound peptide was washed off with PBS-T (PBS + 0.1% Tween20) and samples were incubated with 1 μM heparin in PBS. After another washing step, scaffolds were blocked with 10% BSA in PBS for 30 min and incubated with 1 μM CXCL12 or 0.1 μM FGF-2 or VEGF in PBS for 2 h. Scaffolds were again washed with PBS-T following extensive rinsing with PBS to remove Tween20.2.6 Cell culture
Human umbilical vein endothelial cells (HUVEC, pooled donors; Lonza # CC-2519 (contamination excluded by STR profiling)) were cultured at 37 °C, 5% CO2 and 95% humidity in EGM-2 medium (Lonza) and used between passage 5 and 9. For cell assays, HUVEC were washed with PBS and detached using trypsin/EDTA. After centrifugation at 130 g for 5 min, cells were resuspended in medium without additives (MCDB131) to ensure that the observed effects are derived from the coating rather than unspecifically adsorbed serum proteins. After initial adhesion, medium was again changed to full growth medium (EGM-2).Jurkat cells (DSMZ ACC282 (confirmed by STR profiling)), were kept in RPMI 1640 with 10% FCS at 37 °C in a humidified atmosphere containing 5% CO2.
2.7 Cell adhesion under static conditions
Cell adhesion on PCLLC scaffolds was investigated by fluorescence microscopy and SEM imaging. 80000 HUVEC per scaffold were seeded in medium without additives and incubated for 4 h. Next, scaffolds were washed with PBS twice and adherent cells were fixed with 4% PFA in PBS for 30 min. For SEM, samples were washed again with PBS and dehydrated with increasing concentrations of ethanol (30, 50, 70, 80, 90, 95, 100%). HUVEC cells were imaged using the low vacuum mode of a Quanta FEG scanning electron microscope (FEI Munich). Measurements were carried out with an accelerating beam voltage of 5 kV and an averaged distance of sample to beam of 5 mm at a chamber pressure smaller than 100 Pa. For fluorescence microscopy, nuclei were counterstained with Hoechst 33342 (1:400 in PBS, Sigma-Aldrich) and images captured at 10-fold magnification on an Axio Observer microscope (Zeiss). For investigation of cell morphology, glass-bottom 8-well μ-slides (Ibidi) were coated with 1 μM peptide in 10 mM Tris buffer pH 7.4 and 4 mg ml−1 collagen I (Life Technologies) in 0.02 N acetic acid overnight, respectively. After washing with PBS, 40000 HUVEC per well were seeded in medium without additives and incubated for 4 h. Subsequently, cells were washed and fixed as described above. Permeabilization was carried out with 1% Triton-X100 in PBS, nuclei and F-actin were counterstained with Hoechst 33342 (1:400 in PBS) and TRITC-Phalloidin (1:600 in PBS, Sigma-Aldrich), respectively. Fluorescence microscopy images were taken at 10- and 20-fold magnification. The number of adherent cells was quantified for three 10-fold magnified pictures per well using ImageJ and their size was determined by outlining each cell on three 20-fold magnified pictures using Axio Vision 4.8 (Zeiss). Data was analyzed from three independent experiments performed in quadruplicates and is shown as mean + sem.
2.8 Cell adhesion under flow conditions
Microfluidic chips were fabricated by photopolymerization of polyethylene glycol diacrylate (PEG-DA) using modifications of a procedure previously described for microfluidic chip production.26,27 Briefly, holes at the inlet/outlet positions of the cover plates (Henneberg & Co) were generated using a sand blaster (Barth Serienapparate) with granules of 50 μm particle size. After the TPM coating of the plates, double-sided tape (3M, 468MP 200MP Adhesive) was attached for a defined distance between the glass slides. 200 μl of PEG-DA (Sigma Aldrich, MW 258) with 1% (w/w) 2,2-dimethoxy-2-phenylacetophenone (Sigma Aldrich) was used to fill the space between the glass slides. The chip was placed in the mask aligner (Süss Micro Tec MA6) and a light-impermeable photomask (16000 dpi, Zitzmann GmbH) containing the cell cavity structures was aligned. Photopolymerization was carried out with a Hg lamp (λ = 350–405 nm; P = 12 mW cm−2) at an illumination time of 1.7 s. Subsequently, the nonpolymerized prepolymer was removed by reduced pressure and the microfluidic structures were rinsed with ethanol (70% (v/v) in water) to remove PEG-DA residues. Finally, the chip was re-exposed to UV light for 30 s and, for the connection to pumps, silicon tubes were bonded at the cover plate holes. The cell reservoirs were coated with peptide and collagen I as described earlier. After washing with PBS, reservoirs were filled with a cell suspension of 600000 HUVEC. Following an initial static adhesion time of 3 hours at 37 °C, microfluidic chips were connected to neMESYS syringe pumps (CETONI) fitted out with 30 ml syringes (BD) and cultured with a flow rate of 15 μl min−1 for 16 h at 37 °C. Subsequent fixation and staining was performed analogously to static adhesion studies. Within this experiment, a total of nine microfluidic chips, each coated with the two peptides and collagen I, was assayed. To determine the number of adherent cells, four images with 10-fold magnification were analyzed per cavity, for measuring the cell size, five images with 40-fold magnification were analyzed. Data is represented as mean + sem.
2.9 Cell viability assay
PCLLCNaOH scaffolds were coated with peptides, heparin and cytokines as described earlier and placed into 96-well plates. 40000 HUVEC per scaffold were seeded in medium without additives. After 1 h adhesion, scaffolds were transferred into a new well to exclude cells that had sedimented to the bottom due to scaffold porosity and media was exchanged to EGM-2. After 3 days of cultivation, 20 μl CellTiter-Blue® (Promega) were added and incubated for 2 h. Fluorescence intensity of the supernatant was measured at 560 nm extinction and 590 nm emission (Infinite M200, Tecan). Data was analyzed from five independent experiments performed in triplicates and normalized to untreated scaffolds. Values are represented as mean + sem. For cell adhesion studies, HUVEC cells were incubated in medium without additives for 4 hours. Subsequently, scaffolds were washed with PBS and transferred to clean wells, before fresh medium and CellTiter-Blue® were added. After 30 min incubation, fluorescence intensity was measured as described above. The experiment was performed at least three times in triplicates and is displayed as mean + sem.
2.10 Tube formation assay
HUVEC cells were seeded on coated PCLLCNaOH scaffolds analogously to the viability assay. After 3 days of cultivation, scaffolds were washed with PBS and incubated with Trypsin/EDTA for 5 min. Cells from replicates were pooled and samples were mixed 1:1 with trypan blue (0.5% in PBS), loaded on cell-counting chips and measured at a Spark 10 M plate reader (Tecan) to determine the cell number. In parallel, 10 μl Matrigel (Corning) were pipetted in each well of a μ-slide angiogenesis (Ibidi) and polymerized at 37 °C and 5% CO2 in a humidified atmosphere. Subsequently, 14000 HUVEC per well were reseeded in medium without additives on basement membrane extract and incubated for 6 h. Tube formation was monitored using phase-contrast microscopy. Two images per well were captured using 5-fold magnification and tube length was quantified using Axio Vision 4.8. Data was analyzed from at least three independent experiments performed in quadruplicates and is plotted as mean + sem.
2.11 Gap closure assay
24-well tissue culture plates (TPP) were coated with peptide, heparin and cytokines as described for PCLLC scaffolds. Following PBS-T and PBS, wells were washed with distilled water to remove excess salt and dried on air. 2-well culture-inserts (Ibidi) were placed in the middle of the wells and 40000 HUVEC were seeded in each compartment as described above. After cultivation overnight, culture-inserts were removed and the confluent cell layers rinsed with PBS. Wells were filled with medium and migration monitored by phase-contrast microscopy. Therefore, two images per well with 5-fold magnification were captured at time points 0, 4, 8 and 12 h. The displayed gap size was measured using Axio Vison 4.8. The experiment was performed thrice in duplicates and data is represented as mean ± sem normalized to untreated cells on tissue culture plate (TCP = 1).
2.12 Transwell migration assay
Coated scaffolds were placed in the lower chamber of a 96-well HTS transwell plate (5 μM pore size, Corning). Jurkat cells were spun down for 5 min at 130 g and resuspended in migration medium (RPMI1640 containing 2% FCS). 4 × 105 cells were seeded in the upper chambers, while the lower compartments were filled with migration medium or medium containing 50 nM CXCL12 (positive control). After 0.5, 1 or 2 h incubation at 37 °C in a humidified atmosphere containing 5% CO2, the number of migrated cells was determined at a Spark plate reader as described earlier. Data was analyzed from at least two experiments performed in triplicates and presented as mean + sem.2.13 Hemocompatibility
Whole blood from human donors (agreement with transfusion law §§ 12a and 18, and the directive of the German Bundesärztekammer by the Institute for Transfusion Medicine, University Hospital Leipzig) was obtained in citrate anticoagulant and centrifuged at 20 °C, 200 g for 15 min for separation. The platelet-containing plasma (PCP) was separated from the red blood cells and platelet adhesion was determined analogously to published protocols.28 Briefly, 2/3 of the platelet-poor plasma (PPP) were removed after a second spin at 600 g for 10 min and platelets were washed with platelet washing buffer (140 mM NaCl, 3 mM KCl, 12 mM NaHCO3, 0.4 mM NaH2PO4, 1 mM MgCl2, 0.1% glucose, pH 6.2). Platelets were counted using a Neubauer chamber and resuspended in 15 ml platelet suspension buffer (140 mM NaCl, 3 mM KCl, 12 mM NaHCO3, 0.4 mM NaH2PO4, 2 mM MgCl2, 2 mM CaCl2, 0.1% glucose, pH 7.4 adjusted with 4% HEPES). 300 μl platelet suspension per scaffold was incubated for 1 h at 37 °C. Subsequently, scaffolds were washed five times with PBS and incubated with 300 μl reaction buffer (0.1 M citrate buffer with 5 mM p-nitrophenyl phosphate, 1% TritonX-100, pH 5.0) for 1 h at 37 °C. Addition of 100 μl 2 M NaOH stopped the reaction and absorption was measured at 405 nm. The number of adherent platelets was determined using a linear standard curve from 0–55000 platelets (R2 > 0.99).
For the hemolysis assay, the red blood cells were washed three times with PBS (centrifugation at 11 °C, 2500 g for 5 min). Coated scaffolds were incubated with 4% red blood cells in PBS at 37 °C for 1 h, 1% SDS served as a positive control. Samples were again centrifuged and the absorption of the supernatant measured at 540 nm. Data was analyzed from two independent experiments performed in triplicates and plotted as mean + sem.
All experiments were performed in accordance with the Guidelines of German Transfusion Law §§ 12a and 18 and Directive of the German Bundesärztekammer, and Experiments were approved by the ethics committee at Medical Faculty of Leipzig University (468/17-ek). Informed consents were obtained from human participants of this study.
2.14 Statistical analysis
Data was analyzed using GraphPad Prism 5.03 (GraphPad Software). Statistical significances were determined by one-way ANOVA following Newman–Keuls multiple comparison test or unpaired student's t-test.3. Results
3.1 Mussel-derived surface-binding peptides mediate coating of biodegradable polymeric scaffolds
Catechol chemistry provides a versatile toolbox for the coating of many types of surfaces, including metal oxides, polystyrene, Mica, PTFE and more.10 Amongst, a DOPA-containing peptide derived from the blue mussel was developed, designated MP, which demonstrated excellent coating ability to hydrophilized PCLLC scaffolds (PCLLCNaOH) as well as glass and polystyrene (Fig. 1b and e). For binding studies, two biotin-tagged peptide derivatives were synthesized (Fig. 1a), whereby one was equipped with DOPA (Bio-MP(+)) and the control peptide displayed tyrosine residues instead (Bio-MP(−)). Both peptides were produced in high purity as confirmed by two different RP-HPLC columns. Peptide identity was verified using MALDI-ToF- and ESI-Ion Trap mass spectrometry. The full analytics of the synthesized peptides are displayed in Table 1.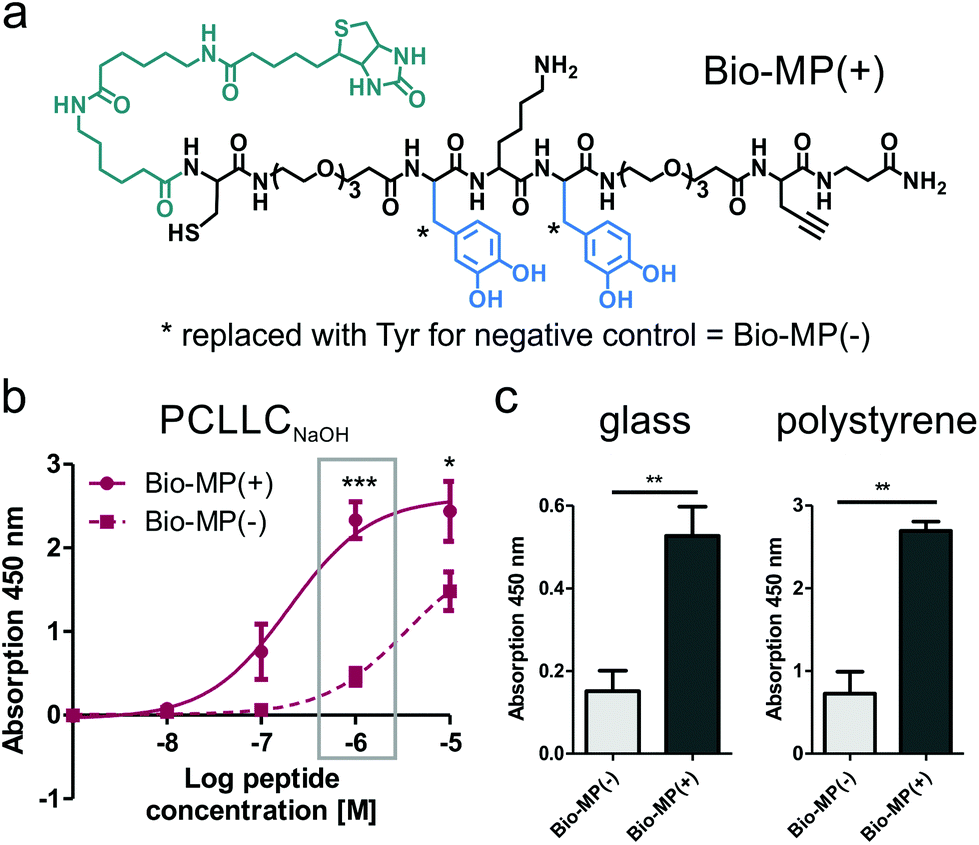 | ||
| Fig. 1 Surface binding properties of mussel-derived peptides. (a) Structure of the DOPA-containing peptide Bio-MP(+), equipped with aminohexanoic acid spacers and a biotin-tag (green) for detection in an ELISA-like assay. The DOPA residues (blue) are replaced with tyrosine to produce the control peptide Bio-MP(−). (b) Mussel-derived peptides bind PCLLCNaOHvia DOPA, shown by an enhanced binding affinity of Bio-MP(+) in comparison to the tyrosine control (Bio-MP(−)). (c) Peptide binding to polystyrene and glass surfaces. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. | ||
| Compound code | Sequence | M calc [Da] | M found [M + H]+ | Elution [% ACN] | Purity [%] |
|---|---|---|---|---|---|
| Peptide identity was confirmed by MALDI-ToF and ESI HCT MS. Purity was quantified from two different HPLC systems. Elution of the peptide was determined from linear gradients of eluent B in eluent A (A: 0.1% TFA in H2O, B: 0.08% TFA in ACN) on a Phenomenex Jupiter Proteo C12 column (90 Å, 4 μm). Bio – biotin, x – L-aminohexanoic acid, EG3 – ethylene glycol, u – L-3,4-dihydroxyphenylalanine, b – L-propargylglycine, β – L-β-alanine, K* – L-ε-azido-lysine. | |||||
| Bio-MP(+) | Bio-xx-C-EG3-uKu-EG3-bβ-NH2 | 1630.6 | 1631.6 | 46 | ≥95 |
| Bio-MP(−) | Bio-xx-C-EG3-YKY-EG3-bβ-NH2 | 1598.9 | 1599.8 | 47 | ≥95 |
| MP | C-EG3-uKu-EG3-bβ-NH2 | 1178.5 | 1179.6 | 36 | ≥95 |
| RGD | c[RGDfK(dienophile)] | 915.5 | 916.5 | 46 | ≥95 |
| HBP | K*-FHRRIKA-NH2 | 1079.4 | 1080.7 | 34 | ≥95 |
| HBP2 | K*-FHRRIKAFHRRIKA-NH2 | 1988.2 | 1989.2 | 29 | ≥95 |
| MP-RGD-HBP | C-EG3-uK(diene(RGD)u-EG3-b(HBP)β-NH2 | 3428.8 | 3429.7 | 31 | ≥95 |
| iMP-RGD-HBP2 | b(HBP2)-EG3-uK(diene(RGD)u-EG3-Cβ-NH2 | 4337.3 | 4338.4 | 34 | ≥95 |
For detection of immobilized peptide, an ELISA-like system was applied. Using a concentration of 1 μM peptide for surface coating, Bio-MP(+) displayed a significantly higher surface binding to PCLLCNaOH with respect to the control peptide Bio-MP(−) (Box in Fig. 1b). Further, binding affinity studies demonstrated significantly higher surface loadings for concentrations above 10−7 M and at least a 17-fold higher affinity with respect to the control Bio-MP(−). Surface binding was also confirmed for polystyrene and glass, where the DOPA peptide was about 3.5-fold enriched with respect to the tyrosine control (Fig. 1c).
Next, different decorations of MP with bioactive peptides were examined for scaffold endothelialization. For integrin binding, a cyclic RGD peptide was chosen, whereas heparin-binding peptides enable interaction with proteoglycans and subsequently also with cytokines (Fig. 2a).
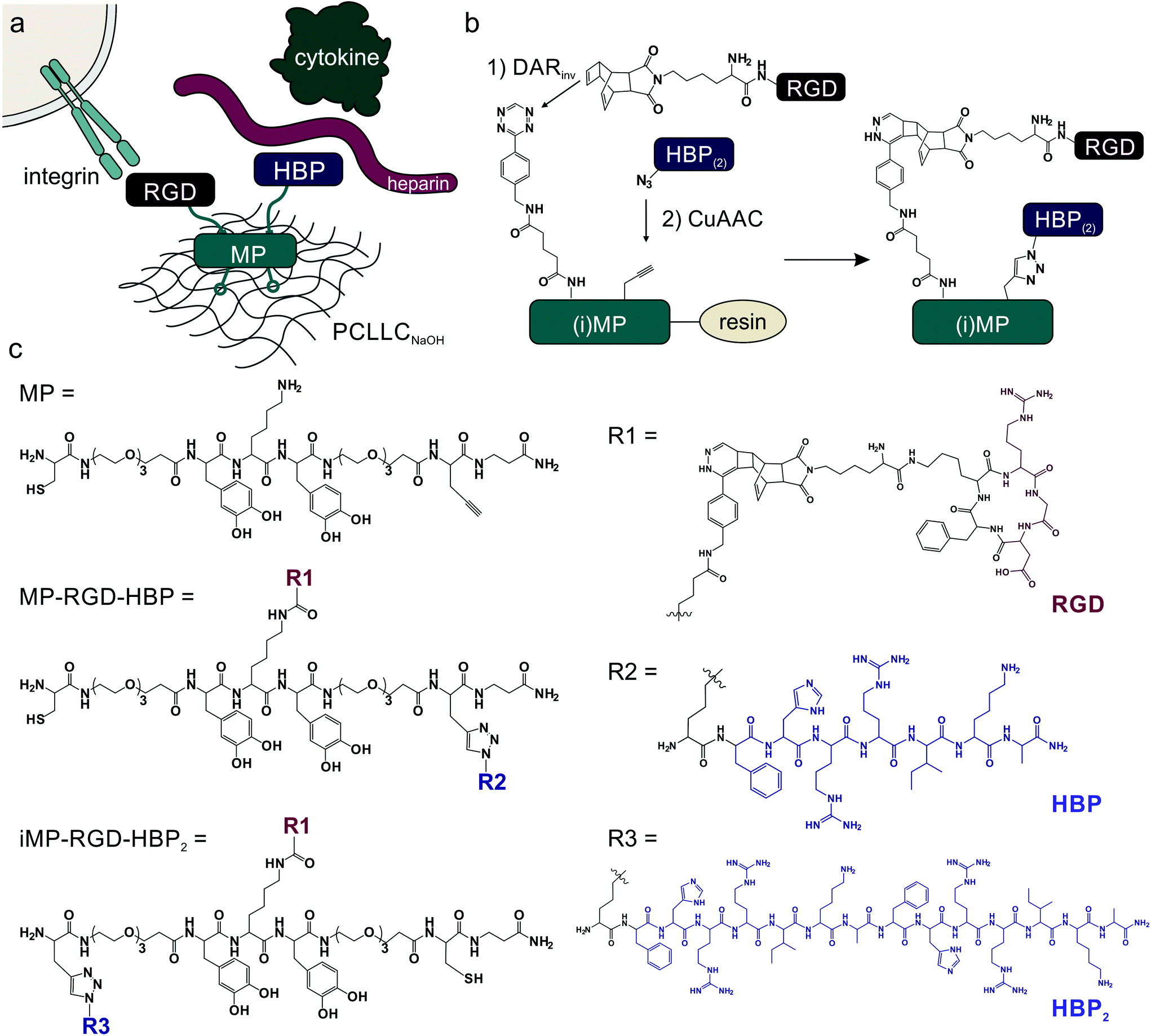 | ||
| Fig. 2 Generation of bioactive peptide coatings. (a) Schematic representation of the multifunctionality of the coating. While MP facilitates the binding to the material, the cyclic RGD peptide mediates cell adhesion and the heparin-binding peptide immobilizes sulfated glycosaminoglycans to subsequently bind and sequester heparin-binding cytokines. (b) A combination of DARinv and CuAAC in an on-resin modification protocol produces functionalized mussel-derived peptides equipped with an integrin- and a heparin-binding peptide. (c) Structural overview of the three tested peptide coatings: MP, MP-RGD-HBP and iMP-RGD-HBP. | ||
For modification with such peptides, the surface-binding peptide MP was equipped with functional groups separated by ethylene glycol spacers. While the lysine side chain was modified with a tetrazine that participates in a Diels–Alder reaction with inverse electron demand (DARinv), the propargylglycine displayed an alkyne functionality for conjugation by copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) (Fig. 2b). The cyclic RGD peptide c[RGDfK] was modified with a dienophile and coupled to the MP in an on-resin modification protocol. Subsequently, a lysine azide-modified heparin-binding peptide, K*(FHRRIKA)n where n = 1 or 2, was coupled by CuAAC. For the longer variant, the position of the propargylglycine in the mussel-derived peptide had to be changed to avoid sterical hindrance, terming the peptide iMP. An overview of the synthesized peptides is given in Fig. 2c, while analytical data confirming peptide identity and purity is displayed in Table 1.
3.2 Functionalization with integrin- and proteoglycan-binding peptides enhances endothelial cell adhesion under static and fluidic conditions
Endothelial cell adhesion to polycaprolactone copolymers is notoriously low, as the polymer itself is very hydrophobic.29 Hydrophilization with NaOH is a known strategy to improve surface compatibility but does not counteract the absence of adhesion motifs that would naturally be presented by the ECM. A positive influence of the adhesion peptides RGD and HBP/HBP2 when immobilized onto these scaffolds was subject to investigation. After 4 h of incubation, adhesion of HUVEC cells to (un)coated PCLLC scaffolds was estimated by measuring the metabolic activity, which correlates to the number of attached cells (Fig. 3a). Herein, cells were seeded in serum-deprived medium to exclude that observed effects are derived from unspecifically-adsorbed serum proteins. As expected, hydrophilization slightly enhanced cell adhesion with respect to unmodified scaffolds. While an additional coating with unmodified MP did not alter adhesion, the functionalized mussel peptides significantly promoted endothelial attachment and increased the metabolic activity once again by 1.5-fold. Fixation and staining of cell nuclei on PCLLC scaffolds revealed only few and scattered cells on untreated surfaces, but extensive adhesion to the MP-RGD-HBP-coated material (Fig. 3b and c).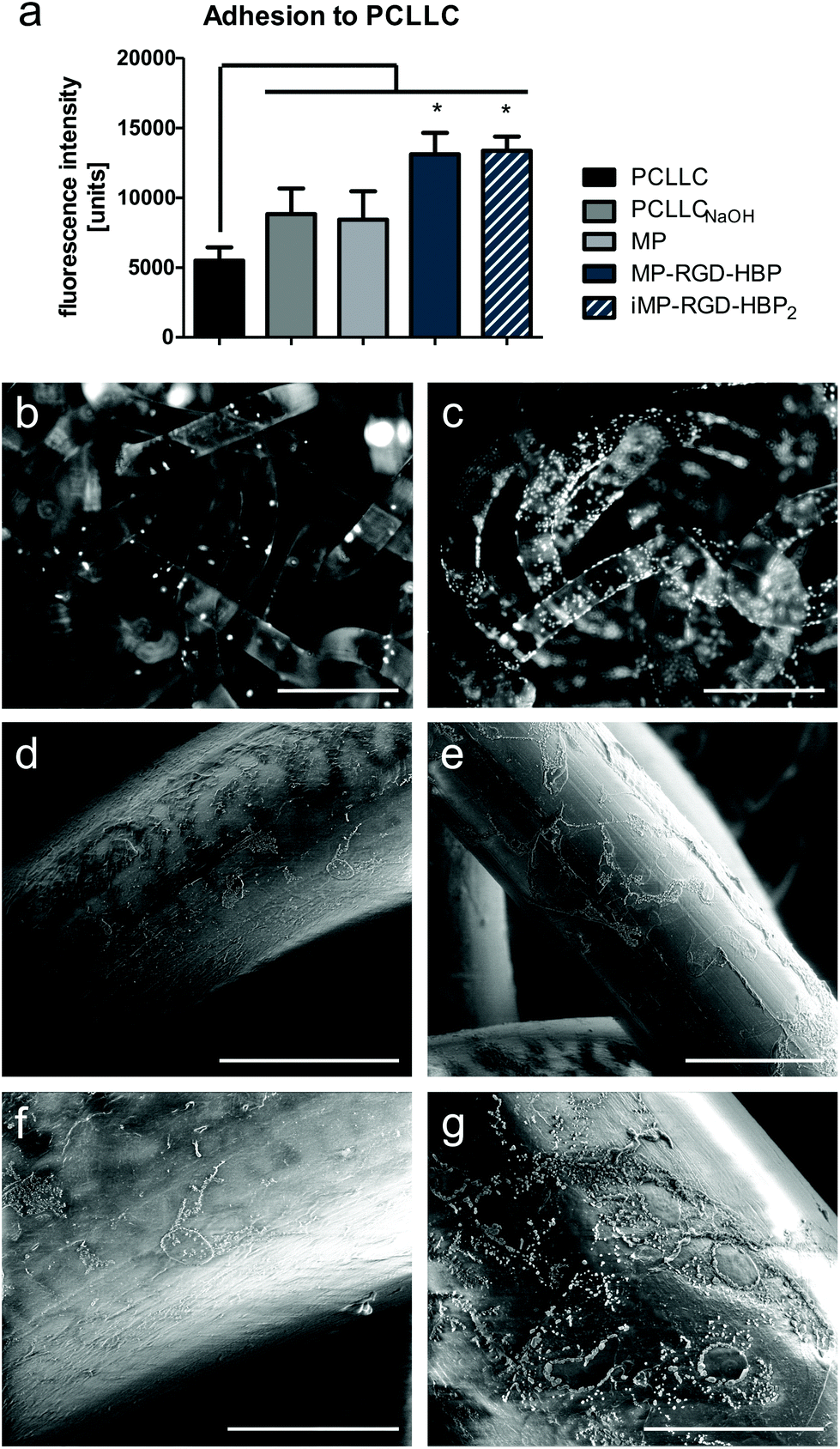 | ||
| Fig. 3 HUVEC adhesion to PCLLC scaffolds after 4 h incubation. (a) Indirect measurement of adherent cells by determination of metabolic activity. (b and c) Fluorescence microscopy images of stained cell nuclei (white dots) on (b) untreated PCLLC and (c) MP-RGD-HBP-coated PCLLCNaOH. Scale bar: 500 μm. (d–g) SEM images of HUVEC on (d and f) untreated PCLLC scaffolds and (e and g) MP-RGD-HBP-coated PCLLCNaOH scaffolds. Scale bars: 100 and 50 μm, respectively. *p ≤ 0.05 with respect to untreated PCLLC. | ||
However, high autofluorescence of the polymer and its 3D structure prevent more detailed investigations of the cell morphology using fluorescence microscopy. Therefore, SEM analysis was performed (Fig. 3d–g). Cells appeared very flat, but deposited salt crystals bordered nuclei and cell membranes, enhancing the visibility of cellular structures. While HUVEC on untreated PCLLC are small and few, MP-RGD-HBP-coated scaffolds promoted cell spreading and attachment.
To analyze the morphology of the endothelial cells in more detail, we switched to coated glass slides. Adherent cells were examined by fluorescence microscopy, enabling the quantification of cell number and size (Fig. 4a–c). While HUVEC were generally found to stay in close proximity with neighboring cells and small connecting filopodia can be observed in all conditions, different cell morphologies distinguished depending on the coating. Cells on MP were much smaller and displayed rather diffuse actin expression while cells on MP-RGD-HBP were far more spread and actin was more organized. On collagen I, the positive control, HUVEC adhered in a more elongated shape with extended filopodia. Quantification revealed that the number of cells that had managed to adhere to the surfaces was indifferent between MP and the collagen I coating, but increased on MP-RGD-HBP and iMP-RGD-HBP2 (Fig. 4g–i). Hereby, iMP-RGD-HBP2 tends to enhance cell adhesion slightly more than MP-RGD-HBP. Quantification of cell size revealed more dramatic differences. Herein, both functionalized MPs significantly increased cell spreading in comparison to MP, while collagen I ranges in between. The growth area covered by adherent HUVEC sums up both observations and displays an increase in the order MP < Col ≪ MP-RGD-HBP = iMP-RGD-HBP2.
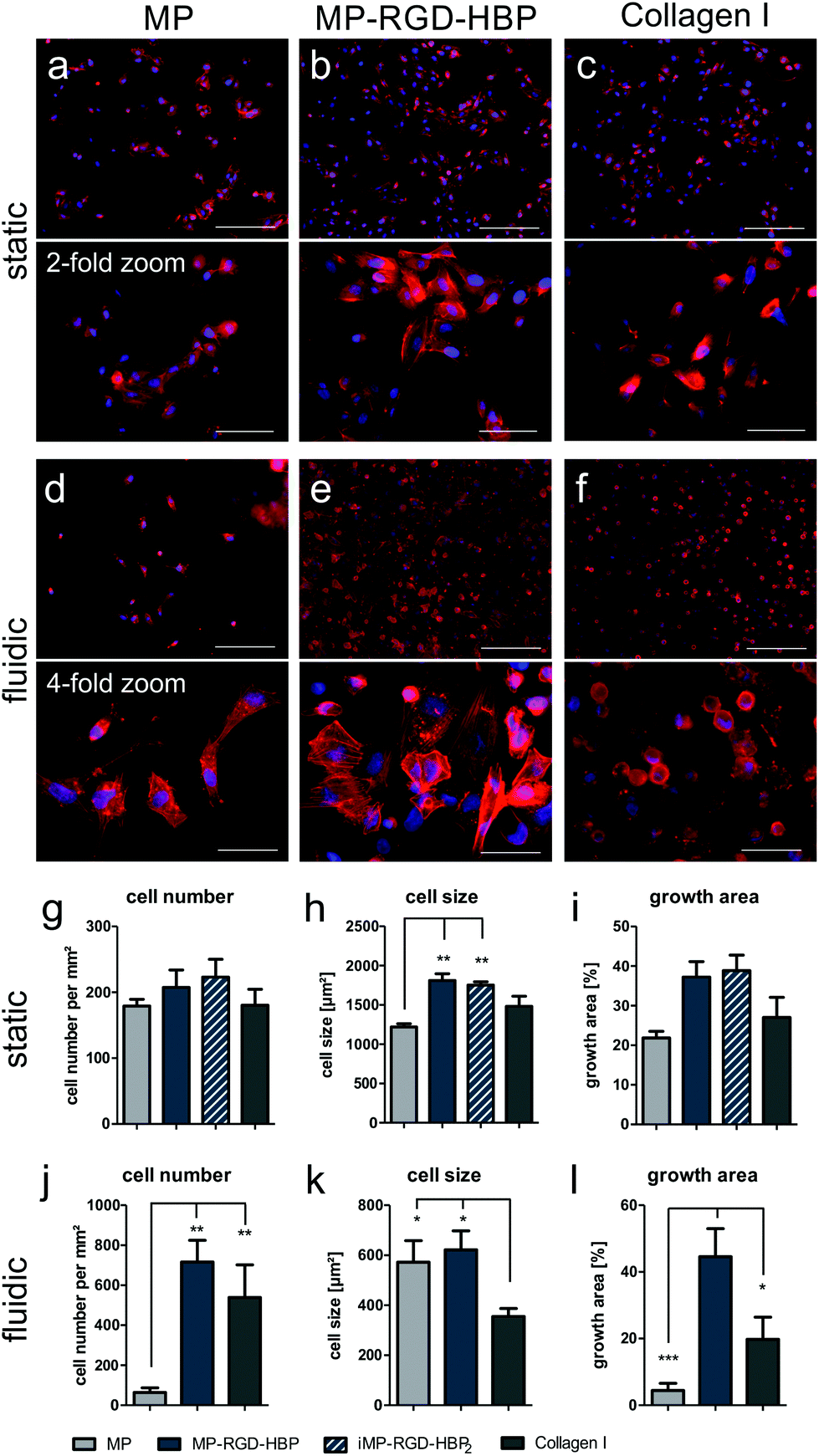 | ||
| Fig. 4 HUVEC adhesion to coated glass surfaces after cultivation under static and fluidic conditions. (a–c) Fluorescence microscopy after 4 h static adhesion to (a) MP, (b) MP-RGD-HBP and (c) collagen I-coated μ-slides. Scale bars: 200 and 100 μm, respectively. (d–f) Fluorescence microscopy after 16 h continuous flow culture on (d) MP, (e) MP-RGD-HBP and (f) collagen I-coated microfluidic cavities. Scale bars: 200 and 50 μm, respectively. The actin cytoskeleton is stained in red, nuclei are stained in blue. (g–i) Quantification of cell size, number of adherent cells and covered growth area after static and (j–l) fluidic culture. *p ≤ 0.05, **p ≤ 0.01. | ||
To approach the in vivo situation wherein endothelial cells are exposed to continuous blood flow and its resulting shear stress, HUVEC were cultivated under fluidic conditions on coated glass surfaces (Fig. 4d–f). On MP, HUVEC could not withstand the pressure very well. After 16 h, many were washed off and cells were only sporadically found. However, those that had managed to attach were mostly spread with a defined actin cytoskeleton. In contrast, the surface was covered much more densely on MP-RGD-HBP than on MP. Most of the cells managed to spread out and produce defined actin stress fibers even though the population remained heterogeneous. On collagen I, almost as many cells could withstand the flow as on MP-RGD-HBP. However, very few HUVEC managed to spread and the far majority displayed a rounded shape. Quantification of the fluorescence microscopy results, displayed in Fig. 4j–l, revealed that the number of adherent cells on MP was indeed significantly lower than on the other two coatings. In contrast, the average cell size differed only marginally between MP and MP-RGD-HBP while being significantly higher than on collagen I. Taken together, the growth area covered by HUVEC cells after exposure to shear flow was lowest on MP, enhanced on collagen I and highest on MP-RGD-HBP, which was significant over both other conditions.
3.3 Delivery of stimulating ECM components can additionally enhance scaffold endothelialization
Besides their interaction properties with cell surface proteoglycans, HBP peptides are likewise suitable for the delivery of heparin and heparin-binding cytokines. Consequential, heparin affinity and the ability to subsequently immobilize the wound-healing cytokines CXCL12, FGF-2 and VEGF onto peptide-coated PCLLCNaOH scaffolds was evaluated in an ELISA (Fig. 5). Fig. 5a demonstrates a strong enhancement of heparin on both functionalized peptides, whereas the elongated HBP2 could bind significantly more heparin than its shorter version. Accordingly, the stepwise increase in bound cytokine continued on MP-RGD-HBP and iMP-RGD-HBP2 (Fig. 5b–d). MP had none or even a protein-repellent effect, as seen for VEGF.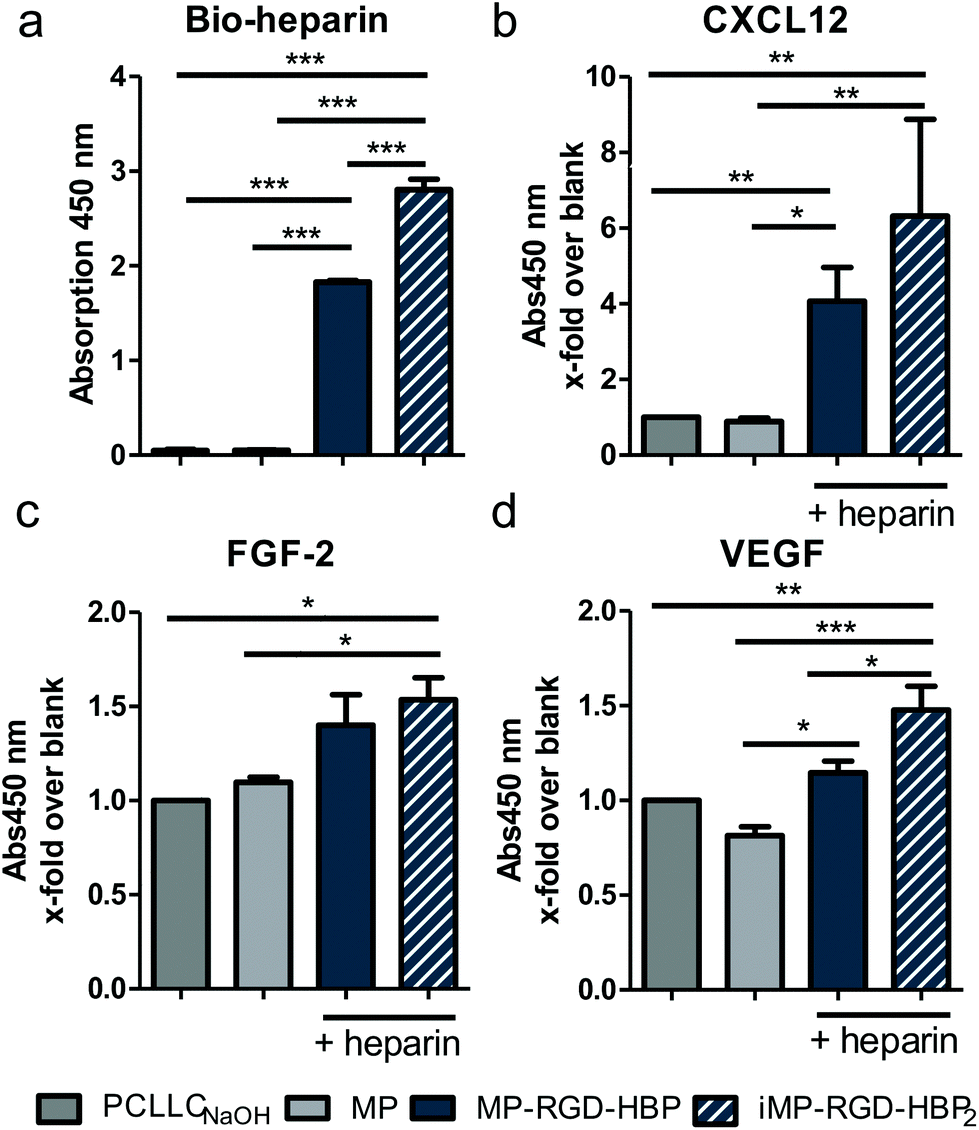 | ||
| Fig. 5 Immobilization of ECM components. (a) Biotinylated heparin is bound to MP-RGD-HBP and iMP-RGD-HBP2-coated PCLLCNaOH scaffolds (b–d) Cytokine immobilization to heparin-loaded MP-RGD-HBP and iMP-RGD-HBP2-coated scaffolds as examined by surface ELISA. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. | ||
To test whether the delivered cytokines in combination with heparin and the peptides positively modulated endothelialization of PCLLC scaffolds, cell viability, differentiation and migration were studied (Fig. 6). Cell viability was measured by metabolic activity three days post-seeding (Fig. 6a). While the slightly positive effect of hydrophilization and MP coating diminished with longer cultivation time, the enhancing effect of the adhesion peptides persuaded. Heparin loading itself and in combination with FGF-2 did not change cell viability, but an additional increase was observed with CXCL12 and VEGF. Cell differentiation was estimated by pseudotube formation on basement membrane extract (Fig. 6b). First, HUVEC were cultivated on the (un)coated scaffolds for three days and then reseeded on Matrigel. When tube length was quantified, scaffold hydrophilization and MP coating proved to be rather obstructive. In contrast, the functionalized MPs MP-RGD-HBP and iMP-RGD-HBP2 tended to enhance average tube length. This was further increased by the delivery of heparin and in combination with the cytokines. Looking at total tube length (Fig. 6c), heparin was again identified as an enhancing factor. Moreover, this effect appeared to be concentration-dependent, as deduced from the different total tube lengths observed with MP-RGD-HBP and iMP-RGD-HBP2. Presentation of cytokines additionally increased network size, especially FGF-2 and VEGF delivered by heparin-loaded MP-RGD-HBP. However, this was not observed on iMP-RGD-HBP2, where total tube length did not exceed the level of heparin.
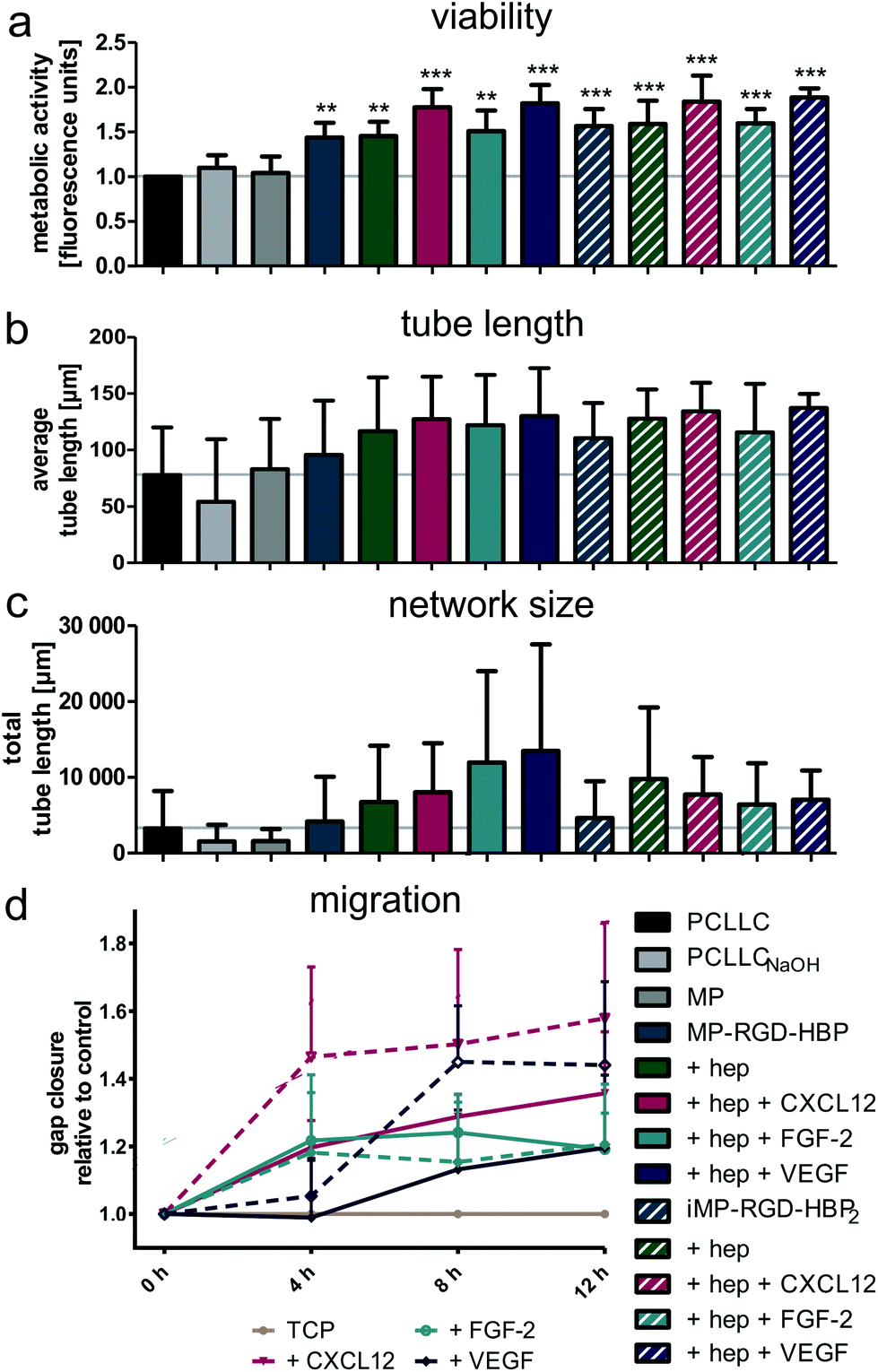 | ||
| Fig. 6 Endothelial response to multifunctional coatings. (a) Cell viability after 3 days cultivation on (un)coated scaffolds. (b and c) Tube formation of HUVEC cells after cultivation on (un)coated PCLLC scaffolds and reseeding on Matrigel. Phase-contrast microscopy images were captured after 6 h incubation and (b) average and (c) total tube length was quantified. (d) Migration of HUVEC cells in response to cytokine stimulation as measured by gap closure assay. Continuous lines refer to coatings on MP-RGD-HBP, dashed to iMP-RGD-HBP2. Hep – heparin, TCP – tissue culture plate. **p ≤ 0.01, ***p ≤ 0.001 with respect to untreated PCLLC. | ||
A possibility for in situ colonization of the scaffolds is the migration of adjacent endothelial cells onto the polymer. Therefore, converging cell sheets in a gap closure assay were monitored microscopically (Fig. 6d). All three cytokines were found to enhance migration within the first 12 h in comparison to the control. The strongest increase was observed with CXCL12 followed by VEGF, which both showed a concentration-dependent effect derived from different protein loadings on MP-RGD-HBP and iMP-RGD-HBP2. Within the first 4 h, FGF-2 increased migration to the same extent as CXCL12, but the curve flattened afterwards. On the contrary, migration towards VEGF was low in the beginning but accelerated between 4 and 8 h.
In addition to the migration of adjacent cells, the attraction and capture of EPCs may contribute to scaffold integration. As CXCL12 is a potent chemoattractant for the recruitment of these cells, its release from coated scaffolds was studied (Fig. 7a). While CXCL12-loaded MP-RGD-HBP + heparin releases about 14 ng ml−1 within the first 24 h, the iMP-RGD-HBP2 coating delivered more than 3-times as much (35 ng ml−1). Next, a transwell migration assay was performed to evaluate whether the chemokine gradient built up by the scaffolds was able to recruit CXCR4-expressing cells (Fig. 7b). Jurkat migration was induced earlier on iMP-RGD-HBP2 coatings, as seen by a small increase after 0.5 h, but both peptide coatings enhanced migration to the same extent within 1 h. Following 2 h of incubation, CXCL12 delivered by MP-RGD-HBP + heparin tends to be even slightly ahead of iMP-RGD-HBP2 + heparin.
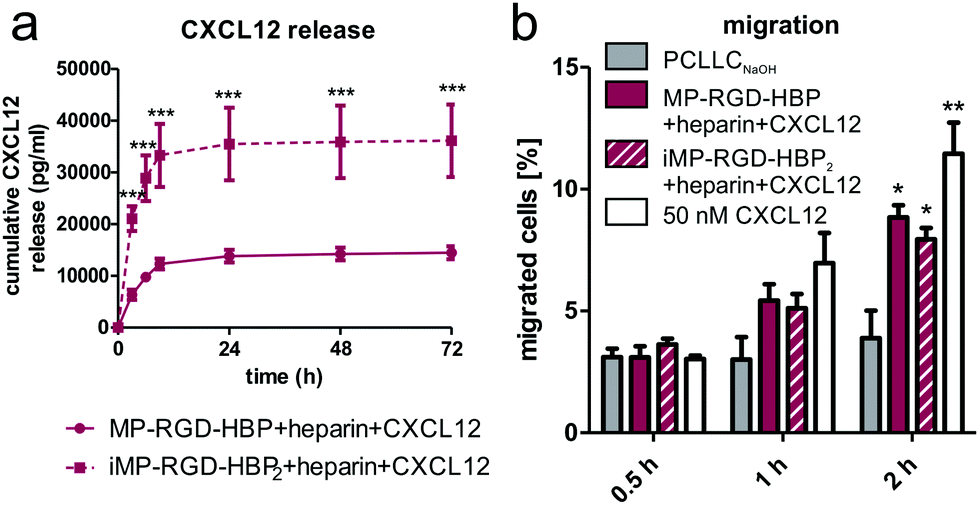 | ||
| Fig. 7 Chemokine release and cell recruitment. (a) Cumulative CXCL12 release from coated PCLLCNaOH scaffolds within 72 h as determined by sandwich ELISA. ***p ≤ 0.001 with respect to MP-RGD-HBP + heparin + CXCL12. (b) Transwell migration assay studying the migration of Jurkat cells towards coated scaffolds after 0.5, 1 and 2 h incubation. *p ≤ 0.05, **p ≤ 0.01 with respect to PCLLCNaOH. | ||
3.4 Hemocompatibility of peptide-heparin-cytokine-complexes on PCLLC scaffolds
Due to the constant contact with blood, cardiovascular implants are at risk of thrombosis resulting in major complications and often implant failure. To ensure hemocompatibility, platelet adhesion and hemolysis were investigated (Fig. 8).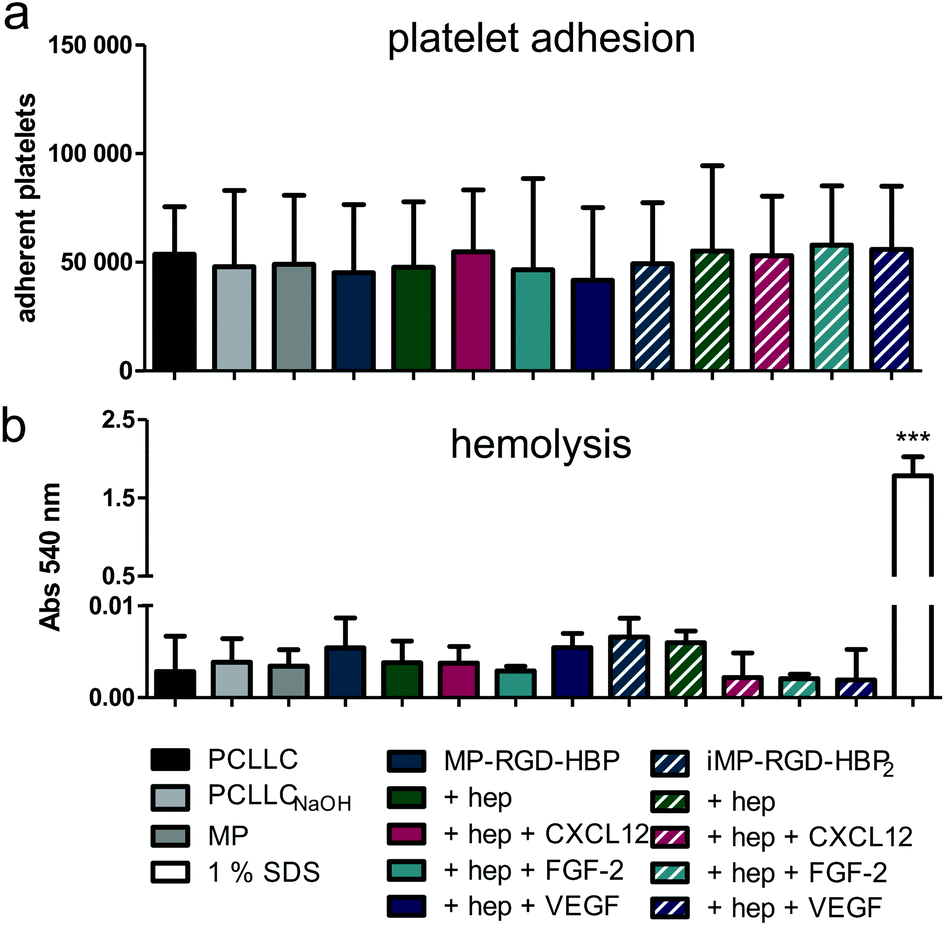 | ||
| Fig. 8 Hemocompatibility of the coatings. (a) Number of adherent platelets after 1 h incubation determined by acid phosphatase activity. (b) Hemolysis of red blood cells after 1 h incubation with coated PCLLC scaffolds in comparison to 1% SDS (positive control). Hep – heparin. ***p ≤ 0.001 with respect to all other conditions. | ||
No enhanced platelet adhesion was observed on any of the coatings with respect to the untreated scaffold (Fig. 8a). Likewise, none of the coatings increased hemolysis to more than 0.4% with respect to the positive control 1% SDS, excluding potential side effects from an extensive lysis of red blood cells (Fig. 8b).
4. Discussion
Insufficient endothelial coverage of cardiovascular implants represents a major risk factor for implant failure due to an enhanced thrombogenicity. To strengthen cell–material-interactions, ECM-inspired coatings are developed that provide endothelial cells with tissue-specific signal molecules modulating cell activation status towards adhesion, proliferation and survival. Peptide-based biomaterial coatings provide a promising toolbox to implement multifunctionality. Coatings combining cell adhesion motifs for integrin and proteoglycan interaction, especially in a spatially-constrained presentation, have shown to synergistically promote cell attachment to surfaces.30,31 Next to functionalization with adhesion peptides, combinations with growth factor-mimetic sequences additionally promote endothelialization.32 However, approaches implementing peptide-mediated and affinity-based incorporation of heparin and cytokines in combination with short adhesion peptides have only been exploited in form of hydrogels so far.33 In the present study, a mussel-derived surface coating combining cell-adhesive as well as heparin-binding and subsequent cytokine-delivering properties is proposed. The basis is comprised of a DOPA-containing surface-binding peptide MP, which is demonstrated here to display a high affinity towards PCLLCNaOH, glass and polystyrene surfaces. Immobilization of MP to PCLLCNaOH is most likely a combination of hydrogen bonding between the hydroxyl groups of the catechol and the oxygen molecules of the polymer as well as dispersion interactions with the phenyl ring. According to literature, a similar binding occurs to glass, where the OH-groups form hydrogen bonds with silanols and siloxanes.34 In contrast, binding to polystyrene is potentially a combination of OH–π, cation–π, including the adjacent lysine residue, and π–π stacking.35–37 Even though the examined materials display apparent differences in surface properties, the binding modes of the DOPA units likewise adapted, which perfectly reflects the versatility of this mussel-derived peptide approach.Initiating a positive healing response from the vascular implant requires modification of the surface-binding peptide MP with bioactives that help guiding the regeneration process. An on-resin modification protocol combining the Diels–Alder reaction with inverse electron demand with the copper(I)-catalyzed azide–alkyne cycloaddition was recently identified as an optimal strategy for the orthogonal introduction of two cell-adhesive peptides.11 The generated bifunctional coatings MP-RGD-HBP and iMP-RGD-HBP2 were tested for their ability to enhance the adhesion of HUVEC cells to PCLLC scaffolds. Both peptides were found to be superior over alkaline hydrophilization, which only provides a generally more favorable interface for unspecific adsorption.38 Coatings with unfunctionalized MP do not compromise endothelial adhesion in comparison to PCLLCNaOH, HUVECs attach equally well owing to its positive surface charge introduced by a free lysine residue. In contrast, cell viability on MP-RGD-HBP and iMP-RGD-HBP2 is significantly enhanced, HUVEC display more organized actin fibers and improved spreading. The presented cyclic RGD peptides specifically address cell surface integrins, promoting the formation of more stable anchor points and linking the surface with the cytoskeleton, causing actin rearrangement.39 Herein, a cooperative action between the proteoglycan-binding HBPs and the RGD peptide is conceivable, as syndecans and integrins synergize to promote focal adhesion contacts and receptor crosstalk.40 While syndecans have a higher potential reach and mediate the initial attachment to distant adhesion ligands,41 integrins finally take their place to form tight interactions necessary for focal adhesion formation.42 Subsequently, adhesion-dependent processes including wound healing and angiogenesis are modulated by the synergistic signaling.43 By trend, iMP-RGD-HBP2 binds more HUVEC cells to coated glass slides than MP-RGD-HBP, possibly due to an enhanced affinity towards heparin and heparan sulfates by HBP sequence elongation.
Under physiologic conditions, exposure to shear stress is a crucial factor in shaping endothelial morphology.44 During microfluidic culture, we observed dramatic changes in HUVEC morphology in comparison to static adhesion studies and the observed differences between coatings intensified. The presentation of cell adhesion peptides by MP-RGD-HBP was found critical for the development of parallel actin stress fibers and a more cobblestone-like cell morphology. This is in agreement with literature, where a combination of integrin- and proteoglycan-binding peptides was identified to induce the alignment and spreading of endothelial cells under flow.31 Notably, collagen I, a standard coating in cell culture, was unable to promote spreading and cytoskeleton organization, even though it likewise displays both binding sites.45 This can be rationalized by the high affinity of the cyclic RGD peptide for integrin αVβ313 which results in an enhanced adhesion strength in comparison to linear sequences and the full-length protein.46 Additionally, anchor strength is enhanced in mussel-derived peptides compared to unspecific protein adsorption. Small peptides combine a number of advantages over structural proteins as they reach a higher packing density, provide receptor selectivity and display ligands in a defined orientation and spacing.47
Nevertheless, dynamic mediators like growth factors, cytokines and chemokines exert substantial roles in orchestrating cell recruitment, proliferation and matrix remodeling during wound healing. Therefore, specific implant coatings should mediate the immobilization and release of such factors. Incorporation of heparin-binding peptides into biomaterial coatings allow the immobilization of heparin and subsequent delivery of heparin-binding cytokines.17 Herein, the interaction between peptide and GAG is based on the electrostatic attraction of negatively-charged sulfate and carboxyl groups of the heparin with basic amino acids within the peptide. Consensus sequences include alternating motifs of basic and hydrophobic amino acids matching the charge density of heparan sulfates.48 Elongating the motif increases the number of potential ionic bonds and can, therefore, retain significantly more heparin. This, in turn, enables an adjustment of the amounts of chemokine bound and released by the implant.
To determine, whether a combination of covalently-attached adhesion molecules with releasable immobilization of heparin and heparin-binding cytokines is beneficial for the integration of biodegradable implants, cell viability and differentiation of HUVEC cultivated on coated PCLLC scaffolds was investigated. After initially elevating cell adhesion to the scaffolds, the functionalized peptides MP-RGD-HBP and iMP-RGD-HBP2 also promoted HUVEC survival. The induced integrin clustering acts as a biosensor for cell–matrix-interactions and mediates a number of anti-apoptotic signals, including PI3-kinase and MAPK pathways.39,49 Additionally, otherwise stimulating growth factors can induce apoptosis in cells lacking appropriate integrin adhesion,49 so the functionalized mussel-derived peptides serve as an indispensable base to fully exploit cytokine stimulation. Required heparin immobilization does not affect endothelial cell viability, verifying the compatibility of this heparin-mediated cytokine delivery approach. On the contrary, heparin co-delivery might have potentiating effects on the pro-angiogenic factors VEGF and FGF-2, as the GAG stabilizes the soluble protein50 and enhances its receptor activity.51,52 Further elevated cell viability was detected on scaffolds coated with peptide, heparin and CXCL12 or VEGF, both known inhibitors of apoptosis and modulators of integrin expression. While CXCL12 promotes anti-apoptotic signaling through its CXCR7 receptor,53,54 it also activates αVβ3 integrins by allosteric modulation.55 Cell surface proteoglycans mediate this interaction, stressing again the interplay between adhesion ligands, GAGs and modulator proteins within the ECM. Combining all of these components in a functional biomaterial coating can induce cooperativity and further enhance the regeneration process. A similar activity is known for VEGF, which likewise inhibits apoptosis56–58 while simultaneously altering integrin expression.59 Co-localization of the VEGF receptor with integrins is even required for full receptor activation.60,61 Despite related effects are known for FGF-2,62 cell viability remained equal to the adhesion peptides. Rationales could be inappropriate loading concentrations or release kinetics of the protein.
Endothelialization of vascular scaffolds technically occurs either by migration from implant edges, transmural migration of endothelial cells or by transformation from EPCs. These can either be recruited from the bone marrow or through newly-formed capillaries.63 Therefore, formation of tube-like structures after cultivation on coated PCLLCNaOH scaffolds was investigated and a stepwise increase in average tube length detected. Presentation of adhesion peptides within MP-RGD-HBP and iMP-RGD-HBP2 was found beneficial, as invasive processes like angiogenesis break cell–cell-contacts and endothelial cells rely more on ECM interactions to sense their surroundings.49 An additional increase in average tube length was promoted by heparin, which is in accordance to literature.64 Further elevation was triggered by stimulation with any of the three cytokines, which are involved in multiple stages of angiogenesis.19 FGF-2 is involved in protease release for matrix remodeling, abolition of cell–cell-contacts, endothelial proliferation and migration,62 while VEGF, a key mediator of angiogenesis,65 guides endothelial sprouting and proliferation.66 CXCL12 stimulates the formation of numerous and long capillary sprouts67 and endothelial branching.68 Results obtained from average tube length measurements were generally transferable to network size. However, total tube lengths on cytokine loaded iMP-RGD-HBP2-heparin-complexes were unexpectedly lower than on MP-RGD-HBP. This could potentially be rationalized from endothelial migration studies, where a concentration-dependent increase was detected for CXCL12 and VEGF. During angiogenesis, migration is upregulated in endothelial cells to encourage sprouting and subsequent connection to other cells. On the contrary, the higher cytokine stimulation might inhibit the formation of stable networks, limiting the observed total tube length. However, higher cytokine release from iMP-RGD-HBP2-heparin-complexes could rather be beneficial for endothelial migration from implant edges. In general, cells at the boundary function as leaders that pass the migratory signal on to the following cell creating a directed sheet migration. If there are no stimulating factors present, as in the control, movements are rather random and more a consequence of backward migration inhibition due to neighboring cells.69 However, VEGF signaling was found to be more potent in higher concentrations when compared to FGF-2, where migration rose earlier but soon reached a lower plateau. Other groups have characterized VEGF as chemotactic, inducing directed cell migration, whereas FGF-2 rather acts as a chemokinetic, generally enhancing cell motility.70 CXCL12 proved to be even more efficient than VEGF in our study. This is in agreement with literature, where CXCL12 is described as one of the most potent chemokines for endothelial cells.71 However, the exact amounts of VEGF and FGF-2 immobilized and released by our coating are not quantitatively determined, so immediate comparisons between cytokines could be misleading.
CXCL12 is also involved in the recruitment of EPCs from the bone marrow.72 Here it acts as a chemoattractant as well as a stimulating factor in EPC differentiation to endothelial cells.73 Therefore, CXCL12 release was quantified and subsequent migration of Jurkat cells was estimated. Although iMP-RGD-HBP2-heparin-CXCL12 seems to exceed the threshold for induction of migration the earliest, the co-delivered elevated amounts of heparin might simultaneously inhibit migration by scavenging the protein from cell surface proteoglycans, which would otherwise mediate receptor activation.74 However, complexation with heparin provides protection against proteolytic decay and could potentially increase CXCL12 half-life and thus range of action.75
All materials in contact with blood must be assessed for hemocompatibility to ensure safe implementation. No increase in platelet adhesion to any of our multifunctional coatings was detected, accounting for the discrimination of integrin αIIbβ3 over αVβ3 integrin by the cyclic RGD peptide.13 Additionally, the hemolysis rate was very low, ensuring implant safety also in combination with peptides, heparin and cytokines.
5. Conclusions
In the current study, we propose a modular assembly of adhesion peptides, heparin and pro-angiogenic factors as suitable biomimetic coatings for cardiovascular devices. While immobilization of the bioactive peptides c[RGDfK] and FHRRIKA by catechol-mediated surface binding improved endothelial adhesion under static and even fluidic conditions, the bifunctional peptide coating proved to be superior over unspecifically-adsorbed adhesion proteins like collagen I. Additionally, integrin signaling stimulated cell survival and differentiation which was further enhanced by CXCL12 and VEGF delivery. Due to the tight interaction between integrins and cytokines, a cooperative effect is suggested, stressing the interplay between all extracellular matrix components comprising adhesion ligands, proteoglycans and signal molecules. Future research could include combined immobilization of CXCL12, VEGF and FGF-2 as their expression is regulated by positive feedback loops and a synergism might additionally boost endothelialization.67,70Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support of the German Research Foundation DFG (TRR67/3, project number 59307082 subproject A04 and grant number FOR 2177, 2652/2-2), the Helmholtz-Institute for Metabolism, Adipositas and Vascular Research (HI-MAG), the European Union and the Free State of Saxony as well as the Research Academy Leipzig. We express our thanks to Hannah Lentschat for performing protein immobilization and cell migration studies. Moreover, we thank Kristin Löbner, Regina Reppich-Sacher and Ronny Müller for technical assistance during cell culture and synthesis. We are grateful to Prof. Dr. med. Stefan Rammelt (TU Dresden, TR67/B05) for providing the PCLLC scaffolds.References
- WHO fact sheets: Cardiovascular diseases (CVDs), https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds), (accessed May 2019).
- Y. Zhu, K. Yang, R. Cheng, Y. Xiang, T. Yuan, Y. Cheng, B. Sarmento and W. Cui, Mater. Today, 2017, 20, 516–529 CrossRef CAS.
- D. G. Rizik and B. B. Padaliya, J. Interv. Cardiol., 2016, 29, 546–548 CrossRef PubMed.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- V. Guarino, G. Gentile, L. Sorrentino and L. Ambrosio, Polycaprolactone: synthesis, properties, and applications. In: Encyclopedia of polymer science and technology, Wiley Interscience, Hoboken, NJ, 2004, pp. 1–36 Search PubMed.
- D. Mondal, M. Griffith and S. S. Venkatraman, Int. J. Polym. Mater. Polym. Biomater., 2016, 65, 255–265 CrossRef CAS.
- S. Pashneh-Tala, S. MacNeil and F. Claeyssens, Tissue Eng., Part B, 2016, 22, 68–100 CrossRef CAS PubMed.
- C. V. C. Bouten, P. Y. W. Dankers, A. Driessen-Mol, S. Pedron, A. M. A. Brizard and F. P. T. Baaijens, Adv. Drug Delivery Rev., 2011, 63, 221–241 CrossRef CAS PubMed.
- J. H. Waite and M. L. Tanzer, Science, 1981, 212, 1038–1040 CrossRef CAS PubMed.
- J. H. Ryu, P. B. Messersmith and H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 7523–7540 CrossRef CAS PubMed.
- M. Pagel, R. Hassert, T. John, K. Braun, M. Wießler, B. Abel and A. G. Beck-Sickinger, Angew. Chem., Int. Ed., 2016, 55, 4826–4830 CrossRef CAS PubMed.
- P. C. Brooks, R. A. Clark and D. A. Cheresh, Science, 1994, 264, 569–571 CrossRef CAS PubMed.
- M. Kantlehner, D. Finsinger, J. Meyer, P. Schaffner, A. Jonczyk, B. Diefenbach, B. Nies and H. Kessler, Angew. Chem., Int. Ed., 1999, 38, 560–562 CrossRef CAS PubMed.
- A. Rezania and K. E. Healy, Biotechnol. Prog., 1999, 15, 19–32 CrossRef CAS PubMed.
- E. I. Oduah, R. J. Linhardt and S. T. Sharfstein, Pharmaceuticals, 2016, 9, 38–49 CrossRef PubMed.
- R. Biran and D. Pond, Adv. Drug Delivery Rev., 2017, 112, 12–23 CrossRef CAS PubMed.
- S. E. Sakiyama-Elbert, Acta Biomater., 2013, 10, 1581–1587 CrossRef PubMed.
- P. Chiodelli, A. Bugatti, C. Urbinati and M. Rusnati, Molecules, 2015, 20, 6342–6388 CrossRef CAS PubMed.
- M. J. Cross and L. Claesson-Welsh, Trends Pharmacol. Sci., 2001, 22, 201–207 CrossRef CAS PubMed.
- J. H. W. Distler, A. Hirth, M. Kurowska-Stolarska, R. E. Gay, S. Gay and O. Distler, Q. J. Nucl. Med., 2003, 47, 149–161 CAS.
- S. Liekens, D. Schols and S. Hatse, Curr. Pharm. Des., 2010, 16, 3903–3920 CrossRef CAS PubMed.
- J. Juarez and L. Bendall, Histol. Histopathol., 2004, 19, 299–309 CAS.
- J.-i. Yamaguchi, K. F. Kusano, O. Masuo, A. Kawamoto, M. Silver, S. Murasawa, M. Bosch-Marce, H. Masuda, D. W. Losordo, J. M. Isner and T. Asahara, Circulation, 2003, 107, 1322–1328 CrossRef CAS PubMed.
- R. Hassert, M. Pagel, Z. Ming, T. Haupl, B. Abel, K. Braun, M. Wiessler and A. G. Beck-Sickinger, Bioconjugate Chem., 2012, 23, 2129–2137 CrossRef CAS.
- B. Rentsch, R. Bernhardt, D. Scharnweber, W. Schneiders, S. Rammelt and C. Rentsch, Biomatter, 2012, 2, 158–165 CrossRef.
- F. D. Zitzmann, H.-G. Jahnke, S. A. Pfeiffer, R. Frank, F. Nitschke, L. Mauritz, B. Abel, D. Belder and A. A. Robitzki, Anal. Chem., 2017, 89, 13550–13558 CrossRef CAS PubMed.
- S. Jezierski, L. Gitlin, S. Nagl and D. Belder, Anal. Bioanal. Chem., 2011, 401, 2651–2656 CrossRef CAS.
- M. Vanickova, J. Suttnar and J. E. Dyr, Platelets, 2006, 17, 470–476 CrossRef CAS.
- N. Siddiqui, S. Asawa, B. Birru, R. Baadhe and S. Rao, Mol. Biotechnol., 2018, 60, 506–532 CrossRef CAS PubMed.
- M. Hoyos-Nogues, E. Falgueras-Batlle, M.-P. Ginebra, J. M. Manero, J. Gil and C. Mas-Moruno, Int. J. Mol. Sci., 2019, 20, 1429 CrossRef CAS.
- F. Karimi, V. J. Thombare, C. A. Hutton, A. J. O'Connor, G. G. Qiao and D. E. Heath, Biomaterials, 2018, 187, 81–92 CrossRef CAS PubMed.
- T. Flora, I. G. de Torre, M. Alonso and J. C. Rodríguez-Cabello, J. Mater. Sci.: Mater. Med., 2019, 30, 30 CrossRef PubMed.
- A. Zieris, S. Prokoph, K. R. Levental, P. B. Welzel, M. Grimmer, U. Freudenberg and C. Werner, Biomaterials, 2010, 31, 7985–7994 CrossRef CAS.
- S. A. Mian, X. Gao, S. Nagase and J. Jang, Theor. Chem. Acc., 2011, 130, 333–339 Search PubMed.
- A. M. Baty, P. K. Leavitt, C. A. Siedlecki, B. J. Tyler, P. A. Suci, R. E. Marchant and G. G. Geesey, Langmuir, 1997, 13, 5702–5710 CrossRef CAS.
- R. Wang and T. Xie, Chem. Commun., 2010, 46, 1341–1343 RSC.
- S. Kim, A. Faghihnejad, Y. Lee, Y. Jho, H. Zeng and D. S. Hwang, J. Mater. Chem. B, 2015, 3, 738–743 RSC.
- S. H. Oh and J. H. Lee, Biomed. Mater., 2013, 8, 14101 CrossRef PubMed.
- R. O. Hynes, Cell, 2002, 110, 673–687 CrossRef CAS PubMed.
- J. A. Roper, R. C. Williamson and M. D. Bass, Curr. Opin. Struct. Biol., 2012, 22, 583–590 CrossRef CAS PubMed.
- S. Weinbaum, J. M. Tarbell and E. R. Damiano, Annu. Rev. Biomed. Eng., 2007, 9, 121–167 CrossRef CAS PubMed.
- J. Takagi, H. P. Erickson and T. A. Springer, Nat. Struct. Biol., 2001, 8, 412–416 CrossRef CAS.
- M. R. Morgan, M. J. Humphries and M. D. Bass, Nat. Rev. Mol. Cell Biol., 2007, 8, 957–969 CrossRef CAS PubMed.
- S. McCue, S. Noria and B. L. Langille, Trends Cardiovasc. Med., 2004, 14, 143–151 CrossRef CAS.
- J. Heino, BioEssays, 2007, 29, 1001–1010 CrossRef CAS PubMed.
- Y. Xiao and G. A. Truskey, Biophys. J., 1996, 71, 2869–2884 CrossRef CAS.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS.
- J. R. Fromm, R. E. Hileman, E. E. Caldwell, J. M. Weiler and R. J. Linhardt, Arch. Biochem. Biophys., 1995, 323, 279–287 CrossRef CAS PubMed.
- D. G. Stupack and D. A. Cheresh, J. Cell Sci., 2002, 115, 3729–3738 CrossRef CAS PubMed.
- R. Flaumenhaft, D. Moscatelli and D. B. Rifkin, J. Cell Biol., 1990, 111, 1651–1659 CrossRef CAS PubMed.
- S. Soker, D. Goldstaub, C. M. Svahn, I. Vlodavsky, B. Z. Levi and G. Neufeld, Biochem. Biophys. Res. Commun., 1994, 203, 1339–1347 CrossRef CAS PubMed.
- D. M. Ornitz, BioEssays, 2000, 22, 108–112 CrossRef CAS PubMed.
- J. Jin, W.-C. Zhao and F. Yuan, Ophthalmic Res., 2013, 50, 6–12 CrossRef CAS PubMed.
- J. M. Burns, B. C. Summers, Y. Wang, A. Melikian, R. Berahovich, Z. Miao, M. E. T. Penfold, M. J. Sunshine, D. R. Littman, C. J. Kuo, K. Wei, B. E. McMaster, K. Wright, M. C. Howard and T. J. Schall, J. Exp. Med., 2006, 203, 2201–2213 CrossRef CAS PubMed.
- M. Fujita, P. Davari, Y. K. Takada and Y. Takada, Biochem. J., 2018, 475, 723–732 CrossRef CAS PubMed.
- I. Spyridopoulos, E. Brogi, M. Kearney, A. B. Sullivan, C. Cetrulo, J. M. Isner and D. W. Losordo, J. Mol. Cell. Cardiol., 1997, 29, 1321–1330 CrossRef CAS PubMed.
- K. Gupta, S. Kshirsagar, W. Li, L. Gui, S. Ramakrishnan, P. Gupta, P. Y. Law and R. P. Hebbel, Exp. Cell Res., 1999, 247, 495–504 CrossRef CAS PubMed.
- J. E. Nör, J. Christensen, D. J. Mooney and P. J. Polverini, Am. J. Pathol., 1999, 154, 375–384 CrossRef.
- K. Suzuma, H. Takagi, A. Otani and Y. Honda, Invest. Ophthalmol. Visual Sci., 1998, 39, 1028–1035 CAS.
- H. Enaida, T. Ito, Y. Oshima, T. Sakamoto, K. Yago, K. Kato and H. Kochi, Fukushima J. Med. Sci., 1998, 44, 43–52 CAS.
- R. Soldi, S. Mitola, M. Strasly, P. Defilippi, G. Tarone and F. Bussolino, EMBO J., 1999, 18, 882–892 CrossRef CAS PubMed.
- M. Presta, P. Dell'Era, S. Mitola, E. Moroni, R. Ronca and M. Rusnati, Cytokine Growth Factor Rev., 2005, 16, 159–178 CrossRef CAS.
- S. Sarkar, K. M. Sales, G. Hamilton and A. M. Seifalian, J. Biomed. Mater. Res., Part B, 2007, 82, 100–108 CrossRef PubMed.
- K. Norrby, Int. J. Exp. Pathol., 2000, 81, 191–198 CrossRef CAS PubMed.
- P. Carmeliet, Oncology, 2005, 69(Suppl. 3), 4–10 CrossRef CAS PubMed.
- H. Gerhardt, M. Golding, M. Fruttiger, C. Ruhrberg, A. Lundkvist, A. Abramsson, M. Jeltsch, C. Mitchell, K. Alitalo, D. Shima and C. Betsholtz, J. Cell Biol., 2003, 161, 1163–1177 CrossRef CAS PubMed.
- F. Mirshahi, J. Pourtau, H. Li, M. Muraine, V. Trochon, E. Legrand, J.-P. Vannier, J. Soria, M. Vasse and C. Soria, Thromb. Res., 2000, 99, 587–594 CrossRef CAS PubMed.
- O. Salvucci, L. Yao, S. Villalba, A. Sajewicz, S. Pittaluga and G. Tosato, Blood, 2002, 99, 2703–2711 CrossRef CAS PubMed.
- U. R. Michaelis, Cell. Mol. Life Sci., 2014, 71, 4131–4148 CrossRef CAS PubMed.
- A. Yoshida, B. Anand-apte and B. R. Zetter, Growth Factors, 1996, 13, 57–64 CrossRef CAS.
- R. Salcedo and J. J. Oppenheim, Microcirculation, 2003, 10, 359–370 CrossRef CAS.
- G. Feng, D. Hao and J. Chai, FEBS J., 2014, 281, 5054–5062 CrossRef CAS PubMed.
- B. Li, W. Bai, P. Sun, B. Zhou, B. Hu and J. Ying, J. Interferon Cytokine Res., 2015, 35, 23–31 CrossRef CAS PubMed.
- G. S. Kuschert, F. Coulin, C. A. Power, A. E. Proudfoot, R. E. Hubbard, A. J. Hoogewerf and T. N. Wells, Biochemistry, 1999, 38, 12959–12968 CrossRef CAS PubMed.
- R. Janssens, S. Struyf and P. Proost, Cell. Mol. Immunol., 2017, 15, 299–311 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01801h |
| This journal is © The Royal Society of Chemistry 2020 |