
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering hemoglobin to enable homogenous PEGylation without modifying protein functionality†
Chris E.
Cooper
 *a,
Gary G. A.
Silkstone
a,
Michelle
Simons
a,
Svetlana
Gretton
a,
Badri S.
Rajagopal
a,
Victoria
Allen-Baume
a,
Natalie
Syrett
a,
Thoufieq
Shaik
a,
Gina
Popa
b,
XiaoBo
Sheng
b,
Matthew
Bird
b,
Ji-Won
Choi
b,
Riccardo
Piano
c,
Luca
Ronda
cde,
Stefano
Bettati
cdef,
Gianluca
Paredi
f,
Andrea
Mozzarelli
defg and
Brandon J.
Reeder
*a
*a,
Gary G. A.
Silkstone
a,
Michelle
Simons
a,
Svetlana
Gretton
a,
Badri S.
Rajagopal
a,
Victoria
Allen-Baume
a,
Natalie
Syrett
a,
Thoufieq
Shaik
a,
Gina
Popa
b,
XiaoBo
Sheng
b,
Matthew
Bird
b,
Ji-Won
Choi
b,
Riccardo
Piano
c,
Luca
Ronda
cde,
Stefano
Bettati
cdef,
Gianluca
Paredi
f,
Andrea
Mozzarelli
defg and
Brandon J.
Reeder
*a
aSchool of Life Sciences, University of Essex, Wivenhoe Park, Colchester, Essex CO4 3SQ, UK. E-mail: ccooper@essex.ac.uk; reedb@essex.ac.uk; Tel: + 44 Tel: (0) 1206 872119
bAbzena, Babraham Research Campus, Babraham, Cambridge, CB22 3AT, UK
cDepartment of Medicine and Surgery, University of Parma, 43124 Parma, Italy
dBiopharmanet-TEC, University of Parma, 43124 Parma, Italy
eInstitute of Biophysics, CNR, 56124 Pisa, Italy
fSITEIA.Parma, University of Parma, 43124 Parma, Italy
gDepartment of Food and Drug, University of Parma, 43124 Parma, Italy
First published on 15th June 2020
Abstract
In order to infuse hemoglobin into the vasculature as an oxygen therapeutic or blood substitute, it is necessary to increase the size of the molecule to enhance vascular retention. This aim can be achieved by PEGylation. However, using non-specific conjugation methods creates heterogenous mixtures and alters protein function. Site-specific PEGylation at the naturally reactive thiol on human hemoglobin (βCys93) alters hemoglobin oxygen binding affinity and increases its autooxidation rate. In order to avoid this issue, new reactive thiol residues were therefore engineered at sites distant to the heme group and the α/β dimer/dimer interface. The two mutants were βCys93Ala/αAla19Cys and βCys93Ala/βAla13Cys. Gel electrophoresis, size exclusion chromatography and mass spectrometry revealed efficient PEGylation at both αAla19Cys and βAla13Cys, with over 80% of the thiols PEGylated in the case of αAla19Cys. For both mutants there was no significant effect on the oxygen affinity or the cooperativity of oxygen binding. PEGylation at αAla19Cys had the additional benefit of decreasing the rates of autoxidation and heme release, properties that have been considered contributory factors to the adverse clinical side effects exhibited by previous hemoglobin based oxygen carriers. PEGylation at αAla19Cys may therefore be a useful component of future clinical products.
Introduction
Infusing extracellular hemoglobin (Hb) into the vasculature has clinical potential as a blood substitute,1 oxygen carrying therapeutic2 or drug delivery agent.3 However, native hemoglobin is in rapid equilibrium between tetrameric (≈64 kDa) and dimeric (≈32 kDa) forms. The dimer is rapidly cleared via the kidneys, both damaging renal function4 and significantly decreasing the plasma lifetime of extracellular Hb.5 Therefore, either chemical or genetic modifications have been used to prevent renal clearance and enhance plasma lifetime.6 The resultant Hemoglobin Based Oxygen Carrier (HBOC) is able to function to transport oxygen in the circulation for several days following infusion.One modification is to mimic the protective effects of the red blood cell via encapsulating Hb into natural or artificial vesicles.7 Alternatively covalent cross-linking of subunits – either using chemical5 or genetic8 means – can increase the minimum molecular size of Hb preventing renal clearance. If the cross-linked protein is large enough it can also decrease unwanted extravasation of the protein into the surrounding tissue, potentially preventing the undesirable scavenging of the intercellular messenger nitric oxide.9 An attractive alternative to these methods is conjugation with large unreactive organic molecules such as poly(ethylene glycol) (PEG). PEG is particularly attractive as it has been shown to decrease immunogenicity and extend the circulatory lifespan of modified proteins10 and is used in many licensed pharmaceutical products.11 PEGylation of human Hb was used in previous HBOC products in clinical trials by Sangart (MP4, Hemospan12), APEX Pharmaceuticals (PHP13) and Baxter Healthcare (rHb2.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14). PEGylation of bovine Hb is used in Sanguinate®,15 a current product being trialed by Prolong Pharmaceuticals.
14). PEGylation of bovine Hb is used in Sanguinate®,15 a current product being trialed by Prolong Pharmaceuticals.
Activated PEGs are able to conjugate to proteins as acylating reagents, alkylating reagents, or thiol-reactive reagents.11 Human Hb has only one reactive sulfhydryl residue per dimer that can be readily PEGylated using standard thiol reactive agents such as maleimide-PEG (MAL-PEG). Modifications at this site (βCys93) increase – albeit only slightly – the oxygen affinity of Hb.16 However, of greater concern they increase the autoxidation rate of oxyhemoglobin (oxyHb) and make Hb more liable to oxidative damage17 and heme loss.18 Modifications at this site (nitrosation, glutathionylation) in vivo have also been suggested to play an important role in a signaling function of Hb that may be important to maintain in a HBOC.19 Reactivity at βCys93 can be maintained in PEGylated Hb by protecting this residue prior to modification at other surface residues.20
Maleimide-PEG conjugation at non cysteine residues is possible via the use of reagents such as 2-iminothiolane to introduce thiol reactivity at terminal amino residues and surface lysines. Such methods of PEGylation have been used successfully in the design of HBOC products tested in animal models (and clinical trials) such as Hemospan,21 Euro-PEG-Hb22 and Sanguinate®.23 However, two issues have arisen. The first is that the final product is very heterogeneous, due to differences in the efficiency at different protein sites of the reactions both creating free thiol residues and/or the subsequent PEGylation at those residues. Heterogenous products also arise when bifunctional activated PEG reagents react at primary amines in Hb as is the case with PHP. HBOCs formed in these ways can have as few as 0 or as many as 14 PEG chains bound.24,25 A heterogeneous product makes it difficult to test and control the reactivity and side reactions of Hb fractions that may be only small components of the product, but could lead to altered reactions in vivo. In this context, it is perhaps noteworthy that preclinical and clinical data led to Biopure modifying their heterogenous cross-linked HBOC products (HS1, HS-2) to create more homogenous products for veterinary (Oxyglobin®) and clinical use (Hemopure®).26
The second problem is that non-specific PEGylation frequently includes residues key to dimer–dimer interactions at the α/β interface. Therefore, these products are frequently dimeric. Although the PEGylated dimers are still too large for rapid renal filtration, they take on some of the properties of the Hb dimer (enhanced autoxidation, high oxygen affinity, no cooperativity). Although this high affinity was marketed as a useful feature in Hemospan,12 this does restrict the ability to modify Hb functions in any new product. PEGylation under controlled conditions in the absence of oxygen favors the T state of Hb, increasing the concentration of tetrameric HBOC,27 but does not completely remove the problem.24
For these reasons it would be useful to generate a HBOC that is derived from homogenous PEGylation at a site that does not perturb the functional properties of Hb. A recombinant Hb carrying a single reactive free surface cysteine at a site remote from the dimer/tetramer interface could possess these desirable qualities. Therefore, this paper tests the efficacy and functional effects of homogenous PEGylation in human Hb at βCys93 (wild type) and two new positions generated by replacing existing alanine residues with cysteine; βAla13Cys is a site homologous to a surface cysteine naturally occurring in feline Hb and αAla19C is a completely novel site on the α subunit.
Results
Fig. 1 shows the sites chosen for mutation to introduce external, reactive cysteine residues suitable for PEGylation at sites remote from the dimer/tetramer interface. PMB reacts with exposed sulfhydryl residues yielding an absorbance change in the UV. For a single exposed residue in the α (as in A12) or β subunit (as in A1 and A13), the reaction would be expected to be complete at a 0.5:1 PMB:Hb ratio (measuring [Hb] on a per heme basis). PMB reactivity (Fig. 2) was therefore consistent with the lack of any exposed residues in A11 and the presence of two exposed residues per Hb tetramer in A1, A12 and A13 i.e one per α or β subunit.
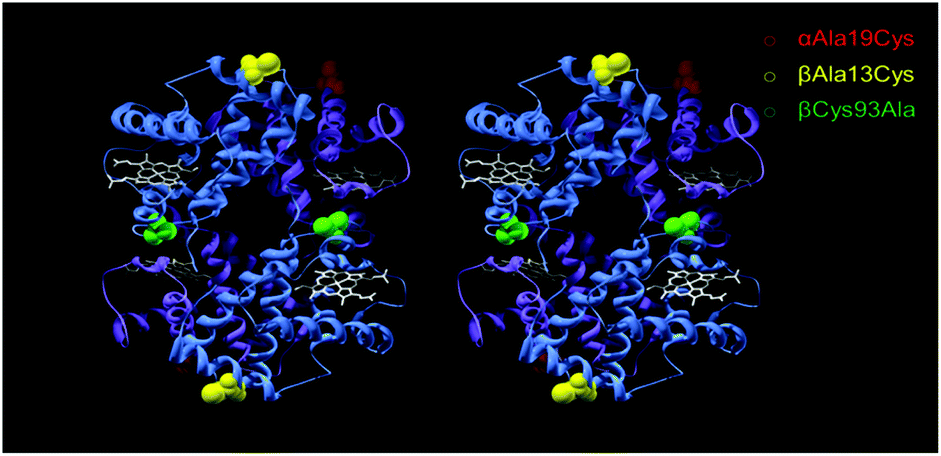 | ||
| Fig. 1 Stereo view of mutation sites for PEGylation. An αAla19Cys and βAla13Cys mutation introduced reactive thiol sites for PEGylation for A12 (red) and A13 (yellow) respectively. For both A12 and A13 the βCys93 site was mutated to an Alanine residue (green). α-Subunit is in purple, β-subunit in blue and heme as white stick. PDB was 1A3N, human hemoglobin (deoxy) mutations were in silico. | ||
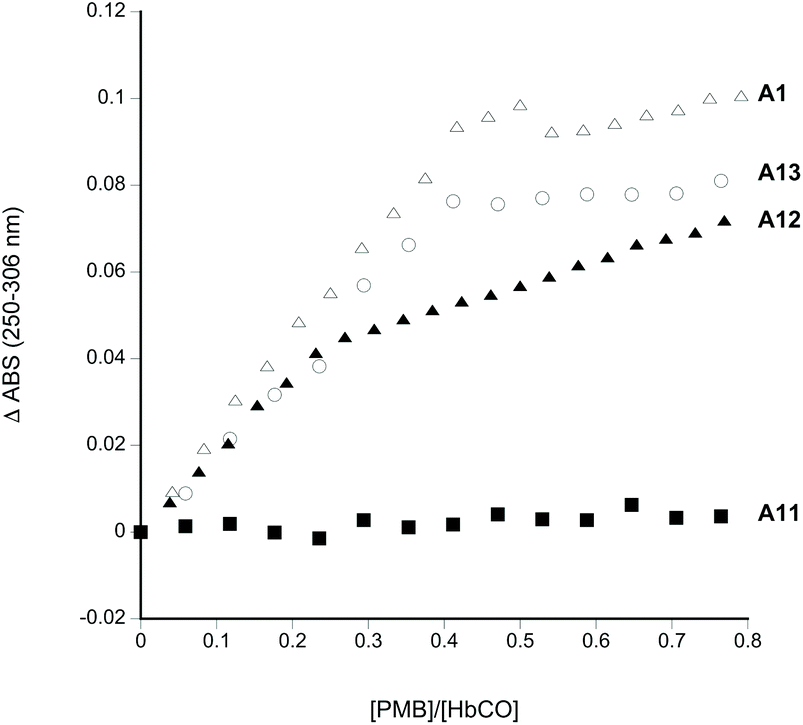 | ||
| Fig. 2 PMB reactivity of Hb mutants. Absorbance change as a function of increasing PMB. Conditions as per materials and methods. Legend: A1, open triangles; A11, filled squares; A12 filled triangles; A13, open circles. | ||
HbCO (R state Hb) was tested to limit undesirable oxidative reactivity during any subsequent PEGylation and purification process. Consistent with previous findings14 there were two reactive thiol residues per α2β2 tetramer for wild type Hb (A1) at βCys93. No reaction was observed when βCys93 was converted to alanine (A11). The creation of new sulfhydryl sites in the α-subunit (A12: βCys93Ala/αAla19Cys) or the β-subunit (A13: βCys93Ala/βAla13Cys) restored this reactivity.
It is important that any new Hb mutations introduced into a putative HBOC do not increase the oxidation of Hb, nor facilitate the release of the heme cofactor. Fig. 3 shows the stability of the recombinant proteins towards autoxidation and heme loss. A12 has lower autoxidation than native Hb. A11 and A12 have lower heme loss than native Hb, whereas A13 has greater heme loss than native Hb. However, in all cases the reactivity is only slightly different to native Hb, indicating that any post translational modifications (or lack of such modifications) in the recombinantly produced proteins exert rather small effects on reactivity in the heme pocket.
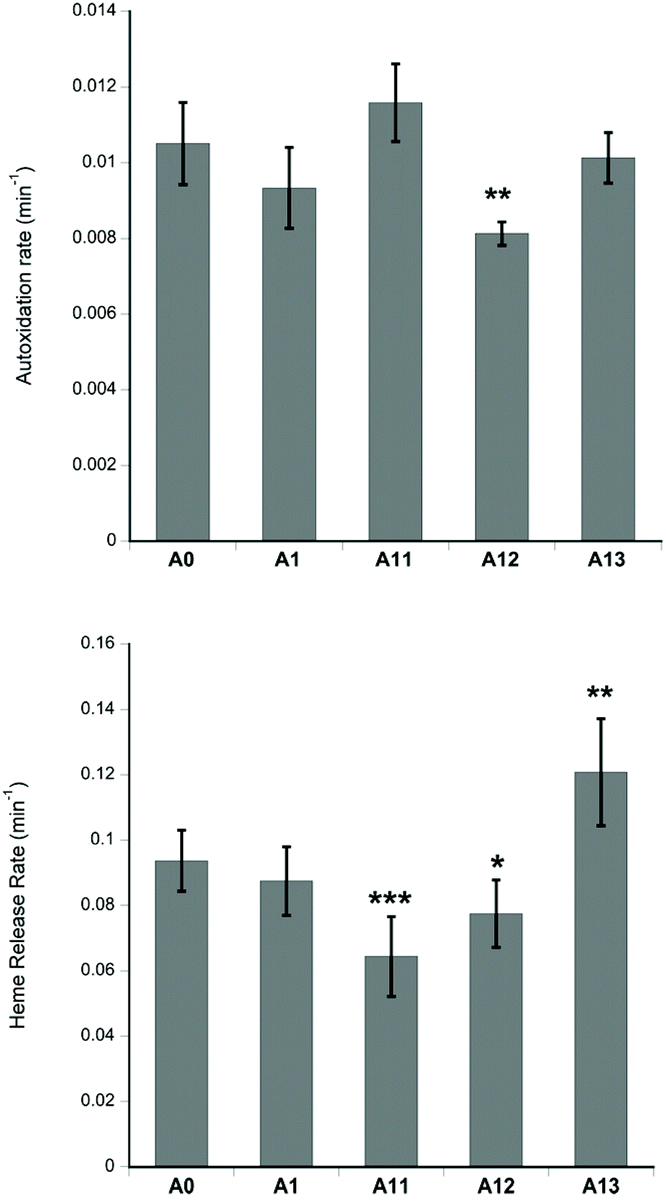 | ||
| Fig. 3 Autoxidation and heme loss. For assay conditions see Materials and methods. Mean ± SD. Statistical significance (using the Student's t-test comparing each recombinant Hb to native human Hb (A0): *p < 0.05, **p < 0.01, ***p < 0.001. | ||
SDS-PAGE (Fig. 4) shows the dominant fraction of the recombinant Hb is consistent with α and β monomers (16 kDa). Consistent with the presence of a free sulfhydryl residue, A1, A12 and A13 are all able to react with 20 kDa MAL-PEG. As seen previously for many proteins,28,29 including Hb,21,24 PEGylated protein adducts run at a slightly higher apparent molecular weight than predicted from unPEGylated protein molecular weight markers. This discrepancy between apparent and real molecular weights in SDS-PAGE of PEGylated Hb can be shown by excising the band and using mass spectrometry.24 Under the conditions of Fig. 4, barium iodide staining showed that the unreacted PEG ran at twice the apparent molecular weight compared to a protein of similar size. So in the case of the Hb monomer (16 kDa) we would expect to observe a mono-20 kDa PEGylated Hb band at ca. 56 kDa (16 kDa for the protein portion plus an apparent 40 kDa for the PEG). The gels in Fig. 4 are therefore consistent with the major new product being Hb (16 kDa) bound to a single (20 kDa) PEG. Note in all cases one subunit will be unreactive to MAL-PEG so that a band remains at 16 kDa – the α-subunit in A1 and A13 and the β-subunit in A12. The visual inspection and the densitometric analysis of gels indicate that A11, A12 and A13 preparations contain a small band at around 30–35 kDa, consistent with an unPEGylated covalent dimer. In some samples of A12 and A13 the proportion of the band in this region increased significantly. In these cases, the excess could be decreased by treatment with dithiothreitol (DTT). However, even post DTT treatment a band remained, suggesting the possibility of a fraction of covalently bound dimer other than a disulfide bridge.
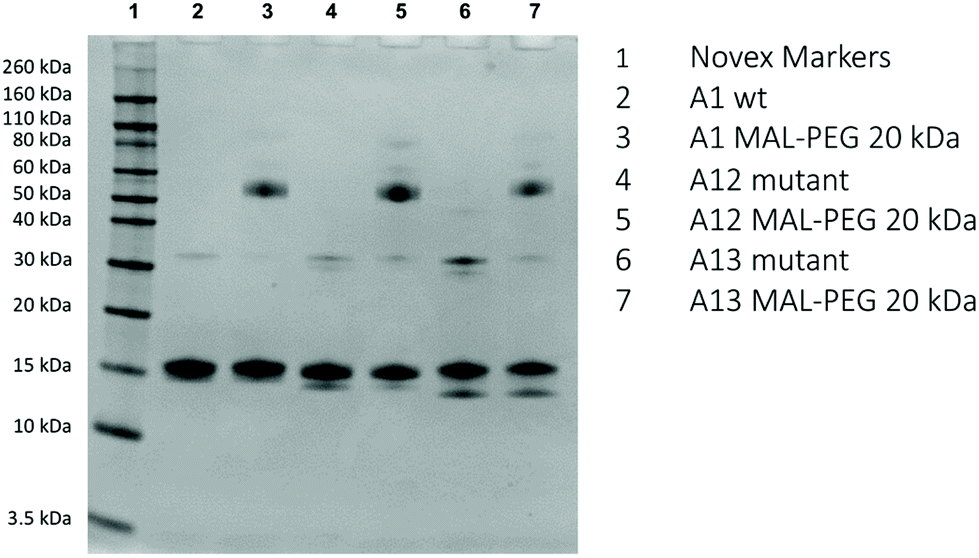 | ||
| Fig. 4 SDS-PAGE before and after PEGylation. For conditions see Materials and methods. | ||
LC-MS QTOF analysis revealed molecular mass for the dominant Hb species both pre and post-DTT treatment of 15289 and 15966, consistent with α and β-subunits with uncleaved N-terminal methionine residues, as is usual for recombinant Hb (see Fig. S1† for A12 data). A minor species observed was more heterogenous, with dominant masses of 30578 and 31932, consistent with the presence of αα and ββ dimers.
Sulfhydryl reactivity and PEGylation of Hb has previously been shown to be partially dependent on the conformational state of the protein (R state or T state). The natural βCys93 site (A1) is more exposed in the R state (favored in oxyHb).30 If a new reactive cysteine was introduced on the surface, distant from parts of the protein that undergo large conformational changes, this differential effect might be absent. This was confirmed in Fig. 5, which shows MAL-PEG reactivity to mutants in R-state Hb (CO-bound) or T-state (deoxygenated). Under the latter conditions, before PEGylation, when hemoglobins were deoxygenated, ascorbate (0.2 mM) was also added as a reducing agent during the reaction. Under both conditions A12 and A13 showed more efficient conjugation than A1 with approximately 50% of the Hb subunits binding PEG (as expected for reactivity at the single external residue in the α- (A12) or β-subunit (A1, A13). Again, no reactivity was seen in the mutant only lacking βCys93 (A11).
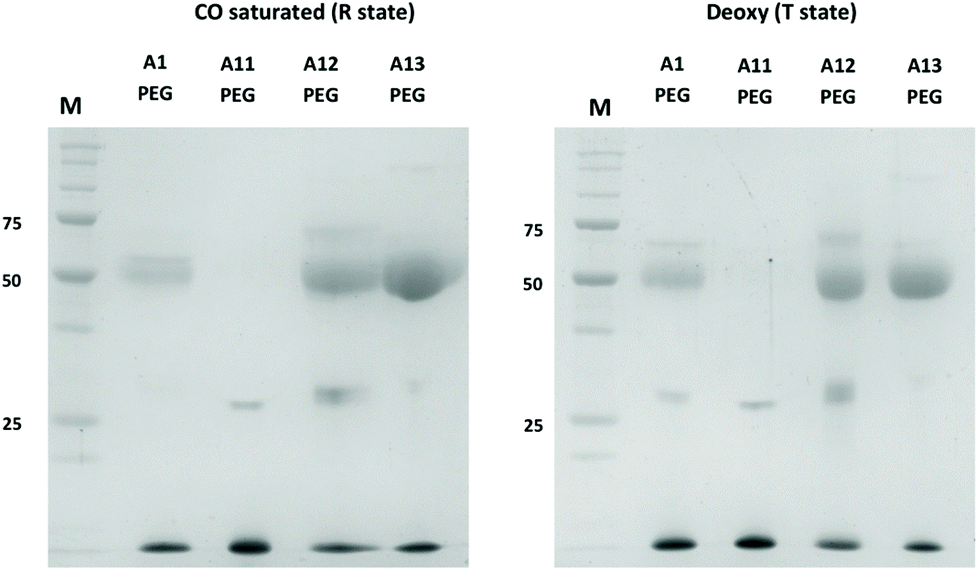 | ||
| Fig. 5 PEGylation efficiency in R-state and T-state Hb. SDS-PAGE of PEGylated Hb A1, A11, A12 and A13 in CO-saturated (left) and anaerobic (right) conditions. Unstained Precision Plus Protein® standards (Biorad) were used as MW markers (25, 50 and 75 kDa bands are highlighted in the gel). For assay conditions see Materials and methods. | ||
It is desirable for PEGylation to be efficient and result in a homogenous HBOC. The extent of PEGylation and the heterogeneity of PEGylated Hb derivatives were evaluated via size exclusion chromatography (SEC) (Fig. 6 and S2†), under conditions where Hb is predominantly dimeric. Results show that PEGylated A12 is more homogenous than PEGylated A13. Moreover, A1 shows a significant amount of unPEGylated protein. The SEC analysis also revealed the presence of a fraction of tetramers for both A12 and A13. This is possibly associated with the reaction of the exposed cysteines and formation of a disulfide bridge between two dimers. However, this did not seem to unduly retard PEGylation efficiency which reveals the order A12 > A13 ≫ A1.
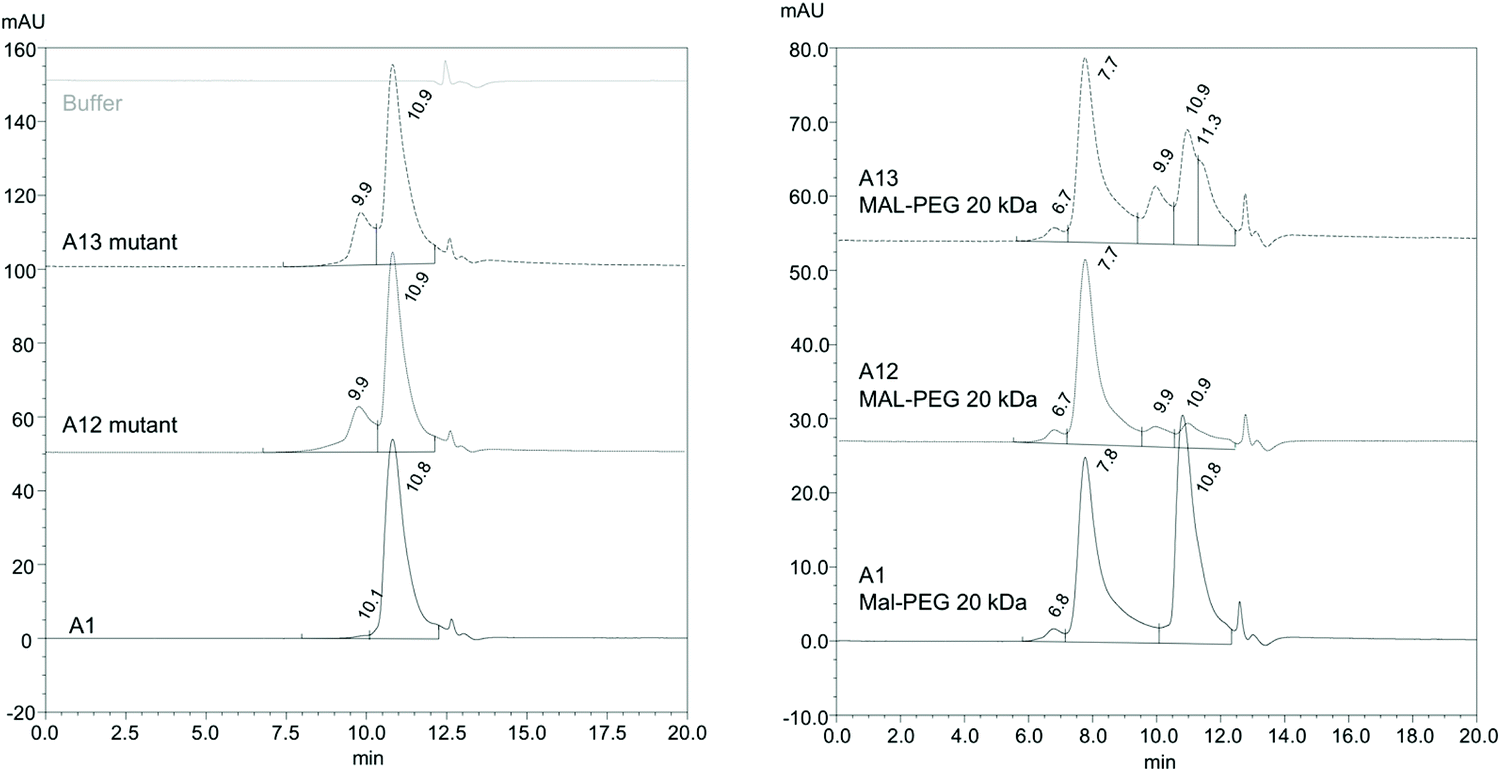 | ||
| Fig. 6 Size exclusion chromatography pre and post PEGylation. SEC of Hb mutants pre (left panel) and post (right panel) PEGylation. For conditions see Materials and methods. For an example of protein MW standards run at the same time as A12 Hb see Fig. S2.† Comparison with SEC standards reveals Hb mutant peaks at 10.8–10.9 min comprise Hb dimer and peaks at 9.9 min tetramer. Hb PEG peaks at 7.7–7.8 min comprise PEGylated dimers. The minor peak at 6.7–6.8 is not straightforward to assign without further analysis. It could be a PEGylated tetramer, but it might also be a different high MW species, e.g. nonspecific protein aggregates or possibly di-PEGylated Hb. | ||
The molecular mass of the MAL-PEG adducts was determined using MALDI/TOF spectrometry (Fig. 7), under oxygenated CO-saturated (oxy) and anaerobic (deoxy) conditions for A12 (Fig. 7A) and A13 (Fig. 7B). In the region 14000–17000 m/z, A12 and A13 showed both α and β peaks, while in the region 30000–34000 three peaks, corresponding to α–α, α–β, and β–β complexes appeared. This is possibly due to artifacts generated by the denaturing conditions used for the mass spectrometry and was also observed for samples of native human HbA0. Upon PEGylation in both R and T state conditions, the A12 sample (Fig. 7A) shows an almost complete disappearance of the peak corresponding to the α chain, in agreement with derivatization of the introduced α Cys19 with 20 kDa MAL-PEG that now exhibits a broad peak around 36000 m/z. Similarly, for A13 (Fig. 7B), there is an almost complete disappearance of the peak corresponding to the β chain, in agreement with derivatization of the introduced β Cys13 with 20 kDa MAL-PEG that exhibits a broad peak around 36000 m/z. It is known that PEG-conjugated proteins exhibit a low propensity to fly and, consequently, they give low signals in mass spectrometers. However, it is clear that the dominant protein product for A12 corresponds to a β-subunit and an α-subunit with a single PEG bound. Likewise, A13 predominantly comprises an α-subunit and a β-subunit with a single PEG bound.
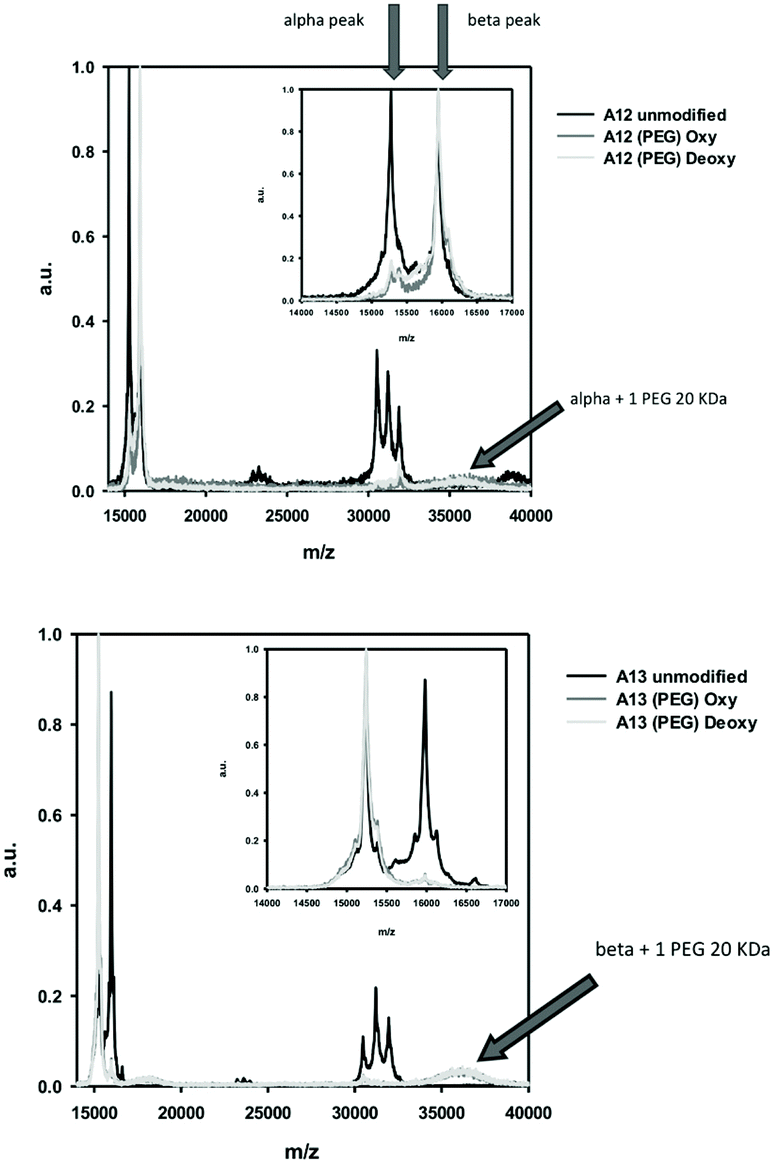 | ||
| Fig. 7 MALDI-TOF of A12 PEG and A13 PEG. Arrows indicate expected positions of α-subunit and β-subunit pre and post PEGylation by a single 20 kDa PEG. Peaks in the 31000–32000 m/z region indicate presence of unPEGylated dimers. | ||
The kinetics of MAL-PEG reactivity was explored in A12 by varying the concentration and time of incubation (Fig. 8). As there are two reactive thiols per tetramer, 100% efficiency with no side reactions or unreacted Hb would have led to 50:50 end ratios of Hb monomer to Hb–PEG adduct. Increasing the PEG:Hb ratio during incubation (from 3:1 from 12:1) and the length of the incubation (from 1–3 h) increased the efficiency of PEGylation in A12. However, the increase was rather small – judged on a per α-subunit basis – from 66% at the lowest PEG ratio (3:1) and shortest incubation time (1 h) to 82% at the highest PEG ratio (12:1) and longest incubation time (3 h).
 | ||
| Fig. 8 Effect of conditions on PEGylation efficiency. SDS-PAGE conditions as in Materials and methods. Lanes indicate different lengths of treatments at different PEG:Hb ratios. Bands were quantified by densitometry and the relative % of total protein stained indicated for each band. | ||
Cooperative oxygen binding is likely to be a useful function in a HBOC and in our mutants, unusually for PEGylated Hb, cooperative oxygen binding was maintained. A12 and A13 PEGylation had no significant effect on oxygen affinity or the Hill coefficient for cooperative binding, although A1 – consistent with previous findings17 – showed a small, but significant, increase in affinity (Table 1 and Fig. S3†).
| P 50 (torr) | P 50 (torr) (after PEG) | Hill coefficient | Hill coefficient (after PEG) | |
|---|---|---|---|---|
| P 50 measured in mmHg. *p < 0.05 compared to pre-PEGylated value. | ||||
| A1 WildType (β-C93) | 5.0 ± 0.3 | 3.8 ± 0.2* | 1.7 ± 0.2 | 1.9 ± 0.2 |
| A13 β-A13C/β-C93A | 4.2 ± 0.2 | 4.6 ± 0.2 | 1.9 ± 0.3 | 2.0 ± 0.2 |
| A12 α-A19C/β-C93A | 4.4 ± 0.2 | 4.1 ± 0.2 | 1.6 ± 0.2 | 1.6 ± 0.1 |
As A12 showed the most efficient PEGylation of our mutants, it was characterized in more detail. PEGylation of A12 resulted in a more stable protein with a decrease in autoxidation and heme loss compared to A1 or unPEGylated A12 (Table 2, Fig. S4 and S5†). Antioxidant reductants such as ascorbate can reduce reactive damaging oxidative Hb species, such as ferryl heme. Accessibility of heme to plasma reductants, such as ascorbate, is therefore desirable to maintain in an HBOC and we tested whether the PEGylation of A12 prevented access of ascorbate to the heme. In wild type Hb, this reaction is fastest in the α subunit. PEGylation on the α-subunit in A12 did not prevent access of ascorbate to the ferryl heme, although there was a decrease in the rate of reduction (decreased Vmax, increased Km). However, there were equivocal effects at the β-subunit (increased Vmax, increased Km).
| A1 WildType (β-C93) | A12 α-A19C/β-C93A | A12-PEG | |
|---|---|---|---|
| *p < 0.05 compared to A12 pre-PEGylated value (determined by Student's t-test for autoxidation and heme release, and via the lack of overlap of 95% confidence intervals of means for non linear curve fitting of ferryl reduction rates). | |||
| Autoxidation | 0.052 ± 0.020 min−1 | 0.048 ± 0.010 min−1 | 0.029 ± 0.003 min−1 * |
| Heme release | 0.046 ± 0.001 min−1 | 0.040 ± 0.010 min−1 | 0.035 ± 0.007 min−1 * |
| Ferryl reduction by ascorbate | |||
| High affinity rate (α subunit) | V max 0.026 ± 0.003 s−1 | V max 0.024 ± 0.001 s−1 | V max 0.014 ± 0.002 s−1 * |
| K M 14.6 ± 5.9 μM | K M 4.32 ± 1.15 μM | K M 18.25 ± 7.87 μM * | |
| Autoreduction rate (α subunit) | 0.0066 s−1 | 0.0040 s−1 | 0.0057 s−1 |
| Low affinity rate (β subunit) | V max 0.015 ± 0.001 s−1 | V max 0.0074 ± 0.0007 s−1 | V max 0.015 ± 0.001 s−1 * |
| K M = 205.7 ± 40.1 μM | K M = 47.1 ± 19.0 μM | K M = 417.2 ± 295.1 μM * | |
| Autoreduction rate (β subunit) | 0.0013 s−1 | 0.0008 s−1 | 0.0009 s−1 |
Discussion
The number of approved biopharmaceuticals incorporating PEG linkages is now well into double figures.11 In most of the current FDA approved proteins, the PEG is introduced in a non site-specific manner resulting in a heterogenous product.31 These products will inevitably include proteins of differing activity and longevity, diminishing the efficacy of the resulting product. Therefore, there is interest in developing site-specific PEGylated products,31 as evidenced by the approvals of human recombinant granulocyte colony-stimulating factor Pegfilgrastim (Neulasta®) and the PEGylated antibody fragment of (TNF)-α monoclonal antibody, certolizamab (Cimzia®). In Pegfilgrastim, site-directed reductive alkylation was used for covalent attachment of PEG to the N-terminal amine group;32 in certolizamab, MAL-PEG is reacted with a C-terminal cysteine in a humanized mouse Fab’ fragment. Our approach uses similar chemistry to certolizamab but instead engineers a target cysteine on the surface of the protein. This is not unique and has been tried in many other proteins, although to our knowledge it is the first time it has been used in Hb.Any introduced site-specific PEG-protein construct must pass three tests: the PEGylation reaction must be efficient; it must not significantly diminish the activity of the protein; and it must not introduce unwanted side reactions. Of the protein we tested A12 (Cys93Ala/αAla19Cys) best fulfills these criteria.
The efficiency of the A12 PEGylation reaction is good (70–80% depending on conditions, Fig. 8). This compares well with the variety of novel surface cysteine reactive residues in granulocyte-macrophage colony-stimulating factor, where efficiencies varies from 45–89%.33 It also compares favorably with the 8–72% yields for the 11 mutations tested in human thyroid stimulating hormone.34 It is also significant that increasing the PEG:Hb ratio above 3:1 and increasing the time of incubation greater than 1 hour had relatively minor effects on this efficiency. This is important, given that grams of PEGylated Hb would be required in any final HBOC product and thus production costs would be a significant fraction of the cost of any commercial product. As a comparison the Hemospan MP4 HBOC was produced with a 20:1 PEG:Hb ratio.21 The natural βCys93 site (A1) is more exposed and reactive in the R state (favored in oxyHb) whereas it is more buried in the T state (favored in deoxyHb).30 However, PEGylation efficiency in A12 is independent of the T or R quaternary state (Fig. 5). This gives additional flexibility in the production of PEGylated A12, which can use either anaerobic or aerobic conditions; in the latter scenario as CO does not hinder PEGylation, it could be added to prevent any oxidation that might occur during processing.
DTT treatment increased the PEG reactivity in some of our preparations, suggesting the presence of disulfide bridges between the introduced cysteine residues. SEC analysis also revealed the presence of a fraction of covalent tetrameric state for both A12 and A13 (Fig. 6). The covalent tetramer could be formed between two dimers of the same Hb molecule, or from two tetramers; these tetramers would then subsequently dissociate into a hybrid tetramer. This has been shown previously with Hb Polytaur (α-Cys104Ser/ α-Ser9Cys human HbA – β-Cys93Ala bovine Hb), although in that case, the formation of a disulfide bridge between two tetramers resulted in the formation of a cyclic trimer of linked tetramers in a time-dependent oxidation process.35 However, disulfide bridges cannot completely explain the small amount of DTT resistant Hb dimer formation that is seen in denaturing SDS-PAGE gels (Fig. 4). It is likely these are due, at least in part, to metal-catalyzed oxidative deamination at the β-subunit36 occurring during the recombinant expression. Consistent with this band excision followed by digestion and peptide mass fingerprinting revealed the presence of predominantly β-subunit. It is clear therefore that, although the A12 mutation appears an ideal candidate for site-specific PEGylation of Hb, more work will be needed to improve the purification process.
Although Hb has a range of activities,37–39 we focused on its primary role in oxygen transport and metabolism. The most exhaustive studies on the effects of site directed mutations on the oxygen affinity of myoglobin (Mb) and Hb have been undertaken by Olson and co-workers.40 They studied over 300 mutants and demonstrated two classes of mutations with significant effects: those that cause local structural changes in the distal heme pocket that change the rate of oxygen entry/exit to/from the heme or alter of the stability of the iron–oxy bond; and those outside the heme pocket near the α1β1 and α2β2 interface that cause global structural perturbations by disrupting the equilibrium between the R (high affinity) and T (low affinity) quaternary states of Hb.41 These active site and allosteric mutations acting independently and in combination have been used to design Hb molecules with P50 values that range from 0.2 to 200 μM, using both allosteric and active site mutations.42 The cysteines we introduced were chosen to be outside the heme pocket and distant from the α1β1 and α2β2 interface, enabling PEGylation that had no effect on both oxygen affinity and cooperative oxygen binding.
To our knowledge the PEGylations on A12 (αAla19Cys) and A13 (βAla13Cys) are the only ones shown to have no significant effect on both oxygen affinity and co-operativity. There is no consensus for the ideal optimum oxygen affinity of an extracellular HBOC. A higher affinity might better deliver oxygen to severely hypoxic tissues, whereas a lower, more physiological, affinity might be better for bulk oxygen transport. These differing strategies are exemplified43 by the higher oxygen affinity (5 Torr) for Sangart's MP4 product, versus values similar to native human Hb for Northfield's Polyheme product (26 Torr) or the lower than native affinities in Biopure's (now HbO2 Therapeutics) Hemopure® product (40 Torr). In this paper we measured oxygen affinities under “pseudophysiological” conditions, similar to those existing outside the red blood cell where high concentrations of physiological effectors such as BPG are not present. So the affinities we measured are lower than those cited above. However, the key fact is that PEGylation of the A12/A13 mutations had no effect on oxygen affinity. These mutations can therefore be used as a template to design other desirable properties without the final PEGylation step modifying oxygen affinity.
As well as mutations specifically designed to modify oxygen affinity,42 other desirable properties that could be added to the A12 template could include adding mutations in the heme pocket to decrease NO scavenging,44 increase ferryl/ferric reduction45,46 or increase nitrite reductase activity.45 Of potential concern is that in order to engineer homogenous PEGylation at a single site we modified the native reactive cysteine residue, β93. The nitrosation of this site has been considered essential for red cell hypoxic signaling,47 though the extent of this effect is controversial.48,49 Our view is that the removal of this reactive residue in extracellular Hb, by replacing it with alanine, is desirable given its tendency to oxidize and cause damage in other HBOCs.50 It is also true that any HBOC infused will still be a minority of the total blood Hb compared to that remaining in the red cell (and which will obviously have intact, reactive βCys93). Therefore, in terms of nitric oxide metabolism we feel the priority in the design of an extracellular HBOC is to decrease the Hb nitric oxide dioxygenase activity and/or increase the nitrite reductase activity. However, we note that, if desired,19 it might be possible to partially protect βCys93 reactivity when PEGylating at α19Cys or β13Cys, depending on the reaction conditions chosen. Clearly this would not be an option if βCys93 itself was chosen as the PEGylation site.
Given its ease of PEGylation and the lack of effect on favorable oxygen binding, we chose to explore further the reactivity of A12 (Cys93Ala/αAla19Cys) in terms of its stability and oxidative reactivity, focusing on heme loss and the autoxidation of ferrous(oxy) Hb to ferric(met)Hb. Heme loss was studied from the ferric Hb as this rate is far greater than the loss from the ferrous redox state51 and is likely to be the physiologically relevant rate for an HBOC in vivo.51,52 The effect of mutations on the rate of globin autoxidation is less well characterized than effects on oxygen affinity.40,42 Although some globins with high oxygen affinity have very low autoxidation rates,53 no Hb mutations that would be able to mirror these effects have been engineered. In general Hb and Mb modifications have no effect or cause an increase in oxidation rate54 and heme loss.42,55 The βCys93Ala/αAla19Cys double mutation itself had no effect on the rate of autoxidation or heme loss, but interestingly, and promisingly, PEGylation at α19Cys decreased both autoxidation and heme loss. This contrasts with PEGylation at βCys93 which elicits the opposite effects.17
It is not easy a priori to predict which surface sites are most efficient for PEGylation in any protein;31 there is always some trial and error involved. However, we conclude that it is indeed possible to engineer an efficient site-specific mutation for PEGylation in human Hb that has no effect on oxygen binding properties and an improvement in the potentially damaging properties of autoxidation and heme loss. PEGylation of Hb at the βCys93Ala/αAla19Cys may therefore prove a useful template as a component of a novel HBOC especially if additional mutations, such as those that decrease nitric oxide scavenging or oxidative stress, are added.
Material and methods
Hb protein purification
Native Hb was purified from human blood as described previously.56 Recombinant DNA fragments encoding the Hb mutations were purchased from Epoch Life Science (Missouri City, Texas, USA). E. coli BL21 (DE3) cells were transformed with pETDuet-1 plasmid containing wild type or mutant genes grown in 2 L Erlenmeyer flasks containing 1.4 L of Luria–Bertani growth medium at 37 °C. The cells were agitated at 180 rpm until an OD600 ∼ 2 was achieved, following which Hb expression was induced by 0.5 mM IPTG (isopropyl β-D-1-thiogalactopyranoside). Also added for enhanced heme synthesis was 0.25 mM ALA (aminolevulinic acid) and 0.1 mM ferric citrate. Cultures were bubbled with pure CO gas. The flasks were sealed thoroughly with rubber bungs and grown for a further 18 h at 30 °C and 90 rpm. Cells were then harvested by centrifugation at 4000 rpm for 20 minutes at 4 °C. Cell pellets were resuspended in 10 mM sodium phosphate buffer, pH 6.0, before being lysed using an Avestin C3 Emulsiflex homogeniser. The samples were kept in the ferrous CO bound form (carbonmonoxyHb, HbCO) throughout the purification process to prevent oxidation. Purification of the Hb protein by cation exchange, anion exchange and affinity chromatography was performed as described previously.57 Samples were stored in liquid nitrogen as the HbCO form and converted to ferrous(oxy) or ferric(met) depending on the reaction measured as described previously.57Protein PEGylation
For specific PEGylation of reactive thiol mutants, PEGylation buffer (100 mM HEPES, 100 mM NaCl, 1.2 mM sodium phosphate, 1 mM EDTA, pH 7) was bubbled with carbon monoxide or nitrogen before adding protein samples. For each sample, absorption spectra were collected in the range of 450–700 nm to check concentration and oxidation state of the proteins: firstly, each sample was diluted with CO-equilibrated buffer (5 μL of stock protein samples + 115 μL of CO-equilibrated buffer) to check the fraction of oxidized Hb. Then, sodium dithionite was added to the sample to reduce oxidized hemoglobin, thus obtaining pure HbCO. Another spectrum was then acquired to determine protein concentration by using a molar extinction coefficient of HbCO of 191 mM−1 cm−1.58 All the samples were brought to 1 mM concentration (on monomer basis) with the CO or nitrogen equilibrated PEGylation buffer for PEGylation in the CO or deoxy forms, respectively. The final volume of the samples to be PEGylated was 200 μL. 20 kDa MAL-PEG (MeO-PEG-mal, Iris Biotech) was added to the protein solutions in 12:1 ratio PEG:Hb tetramer. The samples were left to react at 25 °C for 60 min. MAL-PEG stock solution (10 mM) was prepared from powder just before the reaction. The reaction was quenched by the addition of 2 μL of a 0.9 M Cys solution to each sample. Unreacted reagents and unmodified Hb were removed by ultrafiltration/diafiltration (UF/DF) with 100 kDa MWCO centrifuge filters. A colored flow through was observed for all the samples due to unmodified and/or monoPEGylated Hb. After UF/DF, absorption spectra were collected on the samples to check concentration and oxidation state. The samples were then frozen and stored at −80 °C. PEGylation under anaerobic conditions was carried out in vials fluxed with inert gas, at 25 °C, by adding deoxygenated solutions of reagents using the same protein/reagents ratio used for the CO-saturated samples. Before PEGylation, when hemoglobins were deoxygenated, sodium ascorbate (0.2 mM) was also added as a reducing agent during the reaction.
Cysteine reactivity
Sodium p-hydroxy mercury benzoate (PMB) was freshly prepared prior to each experiment as follows: PMB was dissolved in a small volume of 1 M NaOH. Next, a drop of acetic acid was added. Finally, the PMB solution was cleared by addition of 1 M NaOH drop-wise until the cloudiness was completely gone. Hb solutions were diluted to 20 μM in 70 mM sodium phosphate (pH 7.2) buffer into 1 mL quartz cuvettes and CO was bubbled through the sample. Absorption spectra were recorded after each addition of PMB. Alternative reagents in place of PMB may be used in the measurement of protein sulfhydryls, these include Ellman's reagent and 4,4′-dithiodipyridine.59Oxygen binding measurements
P50 measurements were carried out by diluting the stock Hb solutions in 100 mM HEPES, 100 mM NaCl, 1.2 mM sodium phosphate, 1 mM EDTA, pH 7.0. The final protein concentration was 100 μM (heme basis). Oxygen equilibrium curves were measured at 25 °C, as previously reported.60 For each sample, the absorption spectrum (equilibrated in air) was collected immediately after thawing. Sodium ascorbate and catalase were added to the solution before titrations to reduce metHb and to limit its formation during titration. The samples were deoxygenated using a helium flow and then equilibrated with different oxygen partial pressures. A titration required about 5 hours.SDS-PAGE
The purity of Hb and degree of PEGylation was assessed by 1D-PAGE. SDS-PAGE analyses (Fig. 4 and 8) were performed using NuPAGE 4–12% bis–tris gels and NuPAGE MES SDS running buffer (Invitrogen). Prior to analysis SDS-PAGE samples were prepared by mixing the Hb and PEG–Hb with NuPAGE LDS sample buffer (×4) in a 3:1 ratio. Each analysis was carried out with 1 μg sample loading per well. Electrophoresis was carried out for 35 min at 200 V. Gels were stained using InstantBlue™ (Expedeon) and imaged using an ImageQuant™ LAS4010 system (GE Healthcare). Bands were quantified by densitometry and were sized relative to Novex Sharp Pre-stained Protein Standards (Invitrogen). SDS-PAGE analyses (Fig. 5) were performed using denaturating protein electrophoresis (SDS-PAGE in 12% acrylamide/bisacrylamide), loading 10 μg of unmodified and PEGylated samples. Protein samples were added with sample buffer and brought to 100 °C for 10 minutes. Protein staining was carried out with Biosafe® Coomassie. The SDS-PAGE gels were scanned using a ChemiDoc® imager (Biorad) and bands intensity was evaluated by densitometric analysis.
LC-MS (Q-TOF)
LC-MS analyses were conducted on a Waters NanoAcquity system using an POROSHELL 300SBC3 column (2.1 × 12.5 mm, 5 μm). The mobile phase was composed of aqueous phase (buffer A) and organic phase (buffer B). Mobile phase A was LC-MS grade water containing 0.1% (v/v) formic acid and mobile phase B was LC-MS grade acetonitrile containing 0.1% (v/v) formic acid. The temperature of the column oven was maintained at 60 °C. MS analysis was conducted on a Waters XEVO-G2S Q-TOF mass spectrometer system using electrospray ionisation in positive ion mode (ESI+).Size exclusion chromatography
Size exclusion chromatography (SEC) was carried out using a TOSOH TSKgel Super SW 3000 column (4.6 mm × 30 cm) and guard column (4.6 mm × 3.5 cm). Hb and PEG–Hb sample were isocratically eluted with 200 mM sodium phosphate, pH 6.8 (200 mM KCl, 15% 2-propanol). For each analysis 10 μg of sample was injected and elution was performed over 20 min at 25 °C with UV detection at 280 nm.MALDI TOF mass spectrometry
MALDI TOF mass spectra were acquired in the m/z range 10–40 kDa using a 4800 MALDI TOF/TOF (SCIEX) instrument. Protein samples were mixed with 10 mg mL−1 α-cyano-4-hydroxycinnamic acid (HCCA) matrix solubilized in 75% (v/v) acetonitrile–2.5% (v/v) trifluoracetic acid in 1:9 ratio (v/v) and 1 μL was spotted onto a MALDI TOF plate. Spectra were obtained mediating 500 laser shots in linear positive ion mode.
Autoxidation kinetics (oxyHb to metHb)
The spontaneous autoxidation of 10 μM oxyHb in 70 mM sodium phosphate buffer, pH 7.2, at 37 °C, was monitored optically. Time courses (406–500 nm) of oxy to met conversion were fit to single exponential functions minimizing the least squares using Microsoft Excel™ Solver61 or using the nonlinear curve fitting function in Kaleidagraph™ (Version 4.2). Data are the mean of six experiments.Heme release kinetics
The heme binding scavenger sperm whale (SW) myoglobin (Mb) (H64Y/V67F) was expressed, purified and the heme removed as described previously.62 Sucrose was added to prevent protein precipitation and the reaction followed at lower than physiological pH to increase the rate of heme release and facilitate discrimination between proteins.63 3 μM metHb was incubated with 30 μM apo SW Mb (H64Y/V67F) in 0.15 M sodium acetate, 0.4 M sucrose, pH 5 at 37 °C and reactions monitored using a Tecan Infinite M200 Pro plate reader. The time courses (600–650 nm) for removal of Hb from metHb to apo Mb were fit to a single exponential function minimizing the least squares using Microsoft Excel™ Solver61 or using the nonlinear curve fitting function in Kaleidagraph™ (Version 4.2). Data are the mean of three experiments.Ferryl Hb reduction by ascorbate
Ferryl Hb was made by adding a 3:1 ratio of H2O2:metHb and waiting 10–15 minutes until the spectral change was complete (confirmed by the optical spectra). Trace catalase (1–5 nM) was then added to remove unreacted H2O2. Ferryl reduction (ferryl to met) was then monitored following the addition of varying concentrations of sodium ascorbate (0–1000 μM) to 5 μM ferryl Hb in 70 mM sodium phosphate buffer, pH 7.2. Double exponential fits of the time courses (425–406 nm), assigning the faster phase to the α-subunit and the slower phase to the β-subunit63 were used to calculate the pseudo first order rate constant for ferryl reduction at each [ascorbate]. The autoreduction rate constant at zero [ascorbate] was then subtracted from each rate and the corrected data fit to a rectangular hyperbola using nonlinear regression to determine the Vmax and KM (errors are SEM from the curve fits, n = 12 data points). The program Kaleidagraph™ (Version 4.2) was used for this analysis.
Author contributions
Chris E. Cooper – helped design and plan all studies, wrote paper; Brandon J. Reeder – helped design and plan all studies, edited paper; Luca Ronda, Andrea Mozzarelli, Matthew Bird helped design and plan PEGylation and functional studies, edited paper; Gary G. A. Silkstone, Michelle, Simons, Svetlana Gretton – executed and helped interpret biochemical assays; Badri S. Rajagopal, Victoria Allen-Baume, Natalie Syrett, Thoufieq Shaik – performed protein expression and purification; Gina Popa, XiaoBo Sheng, Ji-Won Choi, Riccardo Piano – helped design, execute and interpret PEGylation, SEC and QTOF studies; Stefano Bettati, Gianluca Paredi – helped design, execute and interpret PEGylation, MALDI TOF MS and functional activity studies.Disclosures
CEC, BJR and GGAS have patents granted and pending relating to modification of hemoglobin amino acids designed to render a blood substitute less toxic and are shareholders in a related company (CymBlood).Abbreviations
| Hb | Hemoglobin |
| Mb | Myoglobin |
| HBOC | Hemoglobin based oxygen carrier |
| A0 | Native human Hb |
| A1 | Wild type recombinant human Hb |
| A11 | Recombinant human Hb with βCys93Ala mutation |
| A12 | Recombinant human Hb with βCys93Ala and αAla19Cys mutations |
| A13 | Recombinant human Hb with βCys93Ala and βAla13Cys mutations |
| metHb | Met(ferric) hemoglobin |
| oxyHb | Oxygenated hemoglobin |
| HbCO | Carbon monoxide bound Hb |
| SW | Sperm whale |
| PEG | Poly(ethylene glycol) |
| MAL-PEG | Maleimide-PEG |
| PHP | Pyridoxalated hemoglobin polyoxyethylene conjugate |
| PMB | Sodium p-hydroxy mercury benzoate |
| SEC | Size exclusion chromatography |
Ethical statement
Blood was donated by adult volunteers in accordance with the University of Essex Guidelines for Ethical Approval of Research Involving Human Participants, and Experiments were approved by the Science and Health Ethics Sub-Committee at the University of Essex. Informed consents were obtained from human participants of this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the UK Medical Research Council (MRC), under grant number MR/L01310X/1 and the UK Biotechnology and Biological Sciences Research Council (BBSRC), under grant number BB/L004232/1.References
- Blood Substitutes, ed. R. M. Winslow, Academic Press, London, 2006 Search PubMed.
- A. T. Kawaguchi, Artif. Organs, 2017, 41, 312–315 CrossRef PubMed.
- S. Brookes, P. Biessels, N. F. Ng, C. Woods, D. N. Bell and G. Adamson, Bioconjugate Chem., 2006, 17, 530–537 CrossRef CAS PubMed.
- M. Feola, J. Simoni, R. Tran and P. C. Canizaro, Biomater., Artif. Cells, Artif. Organs, 1990, 18, 233–249 CrossRef CAS PubMed.
- H. F. Bunn, W. T. Esham and R. W. Bull, J. Exp. Med., 1969, 129, 909–923 CrossRef CAS PubMed.
- Chemistry and Biochemistry of Oxygen Therapeutics: From Transfusion to Artificial Blood, ed. A. Mozzarelli and S. Bettati, John Wiley & Sons, Ltd, 2011 Search PubMed.
- D. R. Harris and A. F. Palmer, Transfus. Altern. Transfus. Med., 2007, 9, 237–245 CrossRef.
- D. Looker, D. Abbott-Brown, P. Cozart, S. Durfee, S. Hoffman, A. J. Mathews, J. Miller-Roehrich, S. Shoemaker, S. Trimble and G. Fermi, et al. , Nature, 1992, 356, 258–260 CrossRef CAS PubMed.
- C. A. Schaer, J. W. Deuel, D. Schildknecht, L. Mahmoudi, I. Garcia-Rubio, C. Owczarek, S. Schauer, R. Kissner, U. Banerjee, A. F. Palmer, D. R. Spahn, D. C. Irwin, F. Vallelian, P. W. Buehler and D. J. Schaer, Am. J. Respir. Crit. Care Med., 2016, 193, 1111–1122 CrossRef CAS PubMed.
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. van Es and F. F. Davis, J. Biol. Chem., 1977, 252, 3582–3586 CAS.
- P. L. Turecek, M. J. Bossard, F. Schoetens and I. A. Ivens, J. Pharm. Sci., 2016, 105, 460–475 CrossRef CAS PubMed.
- K. D. Vandegriff and R. M. Winslow, Artif. Organs, 2009, 33, 133–138 CrossRef PubMed.
- J. L. Vincent, C. T. Privalle, M. Singer, J. A. Lorente, E. Boehm, A. Meier-Hellmann, H. Darius, R. Ferrer, J. M. Sirvent, G. Marx and J. DeAngelo, Crit. Care Med., 2015, 43, 57–64 CrossRef CAS PubMed.
- T. C. Resta, B. R. Walker, M. R. Eichinger and M. P. Doyle, J. Appl. Physiol., 2002, 93, 1327–1336 CrossRef CAS PubMed.
- A. Abuchowski, Artif. Organs, 2017, 41, 346–350 CrossRef PubMed.
- B. N. Manjula, A. Tsai, R. Upadhya, K. Perumalsamy, P. K. Smith, A. Malavalli, K. Vandegriff, R. M. Winslow, M. Intaglietta, M. Prabhakaran, J. M. Friedman and A. S. Acharya, Bioconjugate Chem., 2003, 14, 464–472 CrossRef CAS PubMed.
- T. Hu, D. Li, B. N. Manjula and S. A. Acharya, Biochemistry, 2008, 47, 10981–10990 CrossRef CAS PubMed.
- K. D. Vandegriff, A. Malavalli, C. Minn, E. Jiang, J. Lohman, M. A. Young, M. Samaja and R. M. Winslow, Biochem. J., 2006, 399, 463–471 CrossRef CAS PubMed.
- D. Diesen and J. S. Stamler, J. Mol. Cell. Cardiol., 2007, 42, 921–923 CrossRef CAS PubMed.
- D. Li, T. Hu, B. N. Manjula and S. A. Acharya, Bioconjugate Chem., 2009, 20, 2062–2070 CrossRef CAS PubMed.
- K. D. Vandegriff, A. Malavalli, J. Wooldridge, J. Lohman and R. M. Winslow, Transfusion, 2003, 43, 509–516 CrossRef CAS PubMed.
- E. Alomari, L. Ronda, S. Bruno, G. Paredi, M. Marchetti, S. Bettati, D. Olivari, F. Fumagalli, D. Novelli, G. Ristagno, R. Latini, C. E. Cooper, B. J. Reeder and A. Mozzarelli, Free Radicals Biol. Med., 2018, 124, 299–310 CrossRef CAS PubMed.
- K. Nho, D. Glower, S. Bredehoeft, H. Shankar, R. Shorr and A. Abuchowski, Biomater., Artif. Cells, Immobilization Biotechnol., 1992, 20, 511–524 CrossRef CAS PubMed.
- D. Caccia, L. Ronda, R. Frassi, M. Perrella, E. Del Favero, S. Bruno, B. Pioselli, S. Abbruzzetti, C. Viappiani and A. Mozzarelli, Bioconjugate Chem., 2009, 20, 1356–1366 CrossRef CAS PubMed.
- T. L. Talarico, K. J. Guise and C. J. Stacey, Biochim. Biophys. Acta, 2000, 1476, 53–65 CrossRef CAS.
- J. Y. Chen, M. Scerbo and G. Kramer, Clinics, 2009, 64, 803–813 CrossRef PubMed.
- I. Portoro, L. Kocsis, P. Herman, D. Caccia, M. Perrella, L. Ronda, S. Bruno, S. Bettati, C. Micalella, A. Mozzarelli, A. Varga, M. Vas, K. C. Lowe and A. Eke, Biochim. Biophys. Acta, 2008, 1784, 1402–1409 CrossRef CAS PubMed.
- L. Ronda, M. Marchetti, R. Piano, A. Liuzzi, R. Corsini, R. Percudani and S. Bettati, Pharm. Res., 2017, 34, 1477–1490 CrossRef CAS PubMed.
- Y. Cong, E. Pawlisz, P. Bryant, S. Balan, E. Laurine, R. Tommasi, R. Singh, S. Dubey, K. Peciak, M. Bird, A. Sivasankar, J. Swierkosz, M. Muroni, S. Heidelberger, M. Farys, F. Khayrzad, J. Edwards, G. Badescu, I. Hodgson, C. Heise, S. Somavarapu, J. Liddell, K. Powell, M. Zloh, J. W. Choi, A. Godwin and S. Brocchini, Bioconjugate Chem., 2012, 23, 248–263 CrossRef CAS PubMed.
- Q. H. Gibson, J. Biol. Chem., 1973, 248, 1281–1284 CAS.
- J. K. Dozier and M. D. Distefano, Int. J. Mol. Sci., 2015, 16, 25831–25864 CrossRef CAS PubMed.
- G. Molineux, Curr. Pharm. Des., 2004, 10, 1235–1244 CrossRef CAS PubMed.
- D. H. Doherty, M. S. Rosendahl, D. J. Smith, J. M. Hughes, E. A. Chlipala and G. N. Cox, Bioconjugate Chem., 2005, 16, 1291–1298 CrossRef CAS PubMed.
- H. Qiu, E. Boudanova, A. Park, J. J. Bird, D. M. Honey, C. Zarazinski, B. Greene, J. S. Kingsbury, S. Boucher, J. Pollock, J. M. McPherson and C. Q. Pan, Bioconjugate Chem., 2013, 24, 408–418 CrossRef CAS PubMed.
- S. Faggiano, S. Bruno, L. Ronda, P. Pizzonia, B. Pioselli and A. Mozzarelli, Arch. Biochem. Biophys., 2011, 505, 42–47 CrossRef CAS PubMed.
- J. Levine, M. Weickert, M. Pagratis, J. Etter, A. Mathews, T. Fattor, J. Lippincott and I. Apostol, J. Biol. Chem., 1998, 273, 13037–13046 CrossRef CAS PubMed.
- B. Giardina, I. Messana, R. Scatena and M. Castagnola, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 165–196 CrossRef CAS PubMed.
- J. S. Stamler, L. Jia, J. P. Eu, T. J. McMahon, I. T. Demchenko, J. Bonaventura, K. Gernert and C. A. Piantadosi, Science, 1997, 276, 2034–2037 CrossRef CAS PubMed.
- P. C. Minneci, K. J. Deans, S. Shiva, H. Zhi, S. M. Banks, S. Kern, C. Natanson, S. B. Solomon and M. T. Gladwin, Am. J. Physiol.: Heart Circ. Physiol., 2008, 295, H743–H754 CrossRef CAS PubMed.
- Y. Dou, D. H. Maillett, R. F. Eich and J. S. Olson, Biophys. Chem., 2002, 98, 127–148 CrossRef CAS PubMed.
- D. H. Maillett, V. Simplaceanu, T. J. Shen, N. T. Ho, J. S. Olson and C. Ho, Biochemistry, 2008, 47, 10551–10563 CrossRef CAS PubMed.
- C. L. Varnado, T. L. Mollan, I. Birukou, B. J. Smith, D. P. Henderson and J. S. Olson, Antioxid. Redox Signal., 2013, 18, 2314–2328 CrossRef CAS PubMed.
- T. A. Silverman and R. B. Weiskopf, Transfusion, 2009, 49, 2495–2515 CrossRef PubMed.
- J. S. Olson, E. W. Foley, C. Rogge, A. L. Tsai, M. P. Doyle and D. D. Lemon, Free Radicals Biol. Med., 2004, 36, 685–697 CrossRef CAS PubMed.
- G. G. Silkstone, R. S. Silkstone, M. T. Wilson, M. Simons, L. Bulow, K. Kallberg, K. Ratanasopa, L. Ronda, A. Mozzarelli, B. J. Reeder and C. E. Cooper, Biochem. J., 2016, 473, 3371–3383 CrossRef CAS PubMed.
- C. E. Cooper, G. G. A. Silkstone, M. Simons, B. Rajagopal, N. Syrett, T. Shaik, S. Gretton, E. Welbourn, L. Bulow, N. L. Eriksson, L. Ronda, A. Mozzarelli, A. Eke, D. Mathe and B. J. Reeder, Free Radicals Biol. Med., 2019, 134, 106–118 CrossRef CAS PubMed.
- T. J. McMahon, R. E. Moon, B. P. Luschinger, M. S. Carraway, A. E. Stone, B. W. Stolp, A. J. Gow, J. R. Pawloski, P. Watke, D. J. Singel, C. A. Piantadosi and J. S. Stamler, Nat. Med., 2002, 8, 711–717 CrossRef CAS PubMed.
- R. Zhang, D. T. Hess, J. D. Reynolds and J. S. Stamler, J. Clin. Invest., 2016, 126, 4654–4658 CrossRef PubMed.
- C. W. Sun, J. Yang, A. L. Kleschyov, Z. Zhuge, M. Carlstrom, J. Pernow, N. Wajih, T. S. Isbell, J. Y. Oh, P. Cabrales, A. G. Tsai, T. Townes, D. B. Kim-Shapiro, R. P. Patel and J. O. Lundberg, Circulation, 2019, 139, 2654–2663 CrossRef CAS PubMed.
- Y. Jia, P. W. Buehler, R. A. Boykins, R. M. Venable and A. I. Alayash, J. Biol. Chem., 2007, 282, 4894–4907 CrossRef CAS PubMed.
- T. Kassa, S. Jana, F. Meng and A. I. Alayash, FEBS Open Bio, 2016, 6, 876–884 CrossRef CAS PubMed.
- J. Y. Oh, A. Williams and R. P. Patel, Arch. Biochem. Biophys., 2019, 662, 111–120 CrossRef CAS PubMed.
- A. H. Teh, J. A. Saito, A. Baharuddin, J. R. Tuckerman, J. S. Newhouse, M. Kanbe, E. I. Newhouse, R. A. Rahim, F. Favier, C. Didierjean, E. H. Sousa, M. B. Stott, P. F. Dunfield, G. Gonzalez, M. A. Gilles-Gonzalez, N. Najimudin and M. Alam, FEBS Lett., 2011, 585, 3250–3258 CrossRef CAS PubMed.
- M. F. Tam, N. W. Rice, D. H. Maillett, V. Simplaceanu, N. T. Ho, T. C. Tam, T. J. Shen and C. Ho, J. Biol. Chem., 2013, 288, 25512–25521 CrossRef CAS PubMed.
- E. C. Liong, Y. Dou, E. E. Scott, J. S. Olson and G. N. Phillips Jr., J. Biol. Chem., 2001, 276, 9093–9100 CrossRef CAS PubMed.
- E. M. Welbourn, M. T. Wilson, A. Yusof, M. V. Metodiev and C. E. Cooper, Free Radicals Biol. Med., 2016, 103, 95–106 CrossRef PubMed.
- M. Simons, S. Gretton, G. G. A. Silkstone, B. S. Rajagopal, V. Allen-Baume, N. Syrett, T. Shaik, N. Leiva-Eriksson, L. Ronda, A. Mozzarelli, M. B. Strader, A. I. Alayash, B. J. Reeder and C. E. Cooper, Biosci. Rep., 2018, 38, BSR20180370 CrossRef PubMed.
- E. Antonini and M. Brunori, Hemoglobin and myoglobin and their reactions with ligands, North Holland Publishing Company, Amsterdam, 1971 Search PubMed.
- C. K. Riener, G. Kada and H. J. Gruber, Anal. Bioanal. Chem., 2002, 373, 266–276 CrossRef CAS PubMed.
- L. Ronda, S. Bruno, S. Faggiano, S. Bettati and A. Mozzarelli, Methods Enzymol., 2008, 437, 311–328 CAS.
- D. Fylstra, L. Lasdon, J. Watson and A. Waren, Interfaces, 1998, 28, 29–55 CrossRef.
- G. G. Silkstone, R. S. Silkstone, M. T. Wilson, M. Simons, L. Bulow, K. Kallberg, K. Ratanasopa, L. Ronda, A. Mozzarelli, B. J. Reeder and C. E. Cooper, Biochem. J., 2016, 473, 3371–3383 CrossRef CAS PubMed.
- M. S. Hargrove, E. W. Singleton, M. L. Quillin, L. A. Ortiz, G. N. Phillips Jr., J. S. Olson and A. J. Mathews, J. Biol. Chem., 1994, 269, 4207–4214 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01773a |
| This journal is © The Royal Society of Chemistry 2020 |