
Biomimetic polymeric nanoparticle-based photodynamic immunotherapy and protection against tumor rechallenge
 *a
and
Yu-Kyoung
Oh
*a
*a
and
Yu-Kyoung
Oh
*a
Abstract
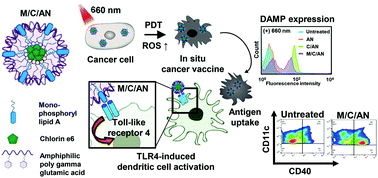
In this study, we sought to design a bionanomaterial that could exert anticancer effects against primary tumors and protect against rechallenged tumors via photodynamic immunotherapy. As a biomaterial, we used an amphiphilic phenylalanine derivative of poly-gamma glutamic acid, which forms nanoparticles by self-assembly. For anticancer effects, we co-entrapped hydrophobic chlorin e6 and monophosphoryl lipid A in the core of the plain amphiphilic phenylalanine nanoparticles (AN), to generate M/C/AN. For comparison, we used plain AN and chlorin e6-loaded AN (C/AN). In vitro studies showed that B16F10 cancer cells treated with C/AN or M/C/AN generated reactive oxygen species and exhibited an enhanced surface display of calreticulin upon exposure to 660 nm light irradiation. C/AN and M/C/AN exerted similar photodynamic anticancer effects; however, M/C/AN, but not C/AN, induced in vitro dendritic cell maturation. Our biodistribution study revealed that C/AN and M/C/AN showed higher accumulation at the tumor tissues compared to that seen in the free chlorin e6-treated group. In B16F10 tumor-bearing mice, the intravenous injection of C/AN or M/C/AN showed similar photodynamic anticancer effects against primary tumors. However, the growth of rechallenged tumors was more significantly inhibited in the M/C/AN group compared to the C/AN group. At day 40 after inoculation of the primary tumor, M/C/AN-treated mice showed 100% survival, whereas the other groups showed 0% survival. In the tumor microenvironment, higher infiltration of CD8+ T cells was observed in the M/C/AN group compared to the other groups. Our results suggest that AN co-loaded with a photosensitizer and an immune stimulant may hold great potential for use in photodynamic immunotherapy to inhibit both primary and metastatic tumors.
- This article is part of the themed collection: Biomimetic Therapeutics
 Please wait while we load your content...
Please wait while we load your content...

