
Clickable, acid labile immunosuppressive prodrugs for in vivo targeting†
Hua
Wang
ab,
Miguel C.
Sobral
ab,
Tracy
Snyder
b,
Yevgeny
Brudno
 ab,
Vijay S.
Gorantla
*c and
David J.
Mooney
*ab
ab,
Vijay S.
Gorantla
*c and
David J.
Mooney
*ab
aHarvard John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, Massachusetts 02138, USA. E-mail: mooneyd@seas.harvard.edu
bWyss Institute for Biologically Inspired Engineering, Cambridge, Massachusetts 02138, USA
cSurgery, Ophthalmology and Bioengineering, Wake Forest School of Medicine, Winston-Salem, NC 27101, USA. E-mail: vgorantl@wakehealth.edu
First published on 30th October 2019
Abstract
Allotransplantation offers the potential to restore the anatomy and function of injured tissues and organs, but typically requires life-long, systemic administration of immunosuppressive drugs to prevent rejection, which can result in serious complications. Targeting the immunosuppressive drug to the graft favors local tissue concentration versus systemic drug exposure and end-organ toxicity. This could reduce the overall dose and dosing frequency of immunosuppressive drugs, and improve the safety and efficacy of treatment. Here, we developed dibenzocyclooctyne (DBCO)-modified prodrugs of the immunosuppressive drugs tacrolimus, rapamycin and mycophenolic acid, and demonstrated their targeted conjugation both in vitro and in vivo to azido-modified hydrogels via Click chemistry. Such azido-modified hydrogels placed in transplanted tissues enable sustained local release of drugs, and could be repeatedly refilled with systemically administered acid-labile prodrugs after drug exhaustion. Thus, clickable prodrugs with degradable linkers provide new possibilities for graft targeted immunosuppression in the context of allotransplantation.
Introduction
The technical, immunologic and functional feasibility of reconstructive transplantation as a promising treatment option for injured tissues has been established in over 200 procedures performed worldwide, including 145 upper extremity (hand), 42 face, and 26 uterus transplants (with over 10 live births).1 Yet, the long-term risks of high-dose or multi-drug systemic immunosuppression remain the key obstacle to life-saving and life-enhancing benefits of such procedures.2,3 Although it has been 65 years since the first kidney transplant was performed by a plastic surgeon, systemic, orally delivered immunosuppression with one or more drugs, remains the standard of care requirement to prevent rejection after transplantation.4,5 The mainstay drug, tacrolimus (FK506, Prograf®), binds to intracellular FKBP-12 receptors and inhibits calcineurin, thus inhibiting interleukin-2 (IL-2) signaling and T cell activation.6 Rapamycin (sirolimus, Rapamune®) suppresses the activation of T and B cells and vascular smooth muscle cell proliferation by inhibiting the mammalian target of rapamycin (mTOR).7–10 Mycophenolate mofetil (MMF, Cellcept®) and its active prodrug mycophenolic acid (MPA), selectively block T-cell proliferation and suppress B-cell antibody production by suppressing guanosine-5′-monophosphate.11–14 Unlike tacrolimus, which is mostly effective in acute rejection, rapamycin and MPA have superior efficacy in chronic rejection.11–13,15–18Orally administered immunosuppressive drugs undergo extensive metabolism in the gastrointestinal tract and liver, greatly reducing their bioavailability. For example, bioavailability of oral tacrolimus is only between 7.3% to 19.7%.19 Taking possible renal clearance into consideration, only a small percentage of drug typically reaches target graft tissues. Thus, drug levels in the bloodstream do not reliably or proportionally correlate with drug concentrations in graft tissues, and acute rejection can occur even when drug levels in the bloodstream are within the “target range”.19–23 For anti-rejection efficacy, large and repeated dosing of immunosuppressive drugs is often necessary, leading to high systemic drug exposure and increased risks of metabolic derangement, infections or malignancy.24–26 Studies have shown that drug levels in the graft tissues are better correlates and monitors of dosing, exposure and efficacy.19,27 Since the graft is the site of the initiation and perpetuation of the innate and adaptive immune response, it may be logical to direct and concentrate the immunosuppressive agents to the graft tissues.
Different from solid organ transplants that are internal, hand or face transplants are externally visible and accessible for diagnostic and treatment interventions, like topical immunosuppression or graft implanted drug delivery systems.28 Graft implanted drug depots could bypass the intestinal/liver metabolism, and enable tunable release of single or multiple drugs over days to weeks. This spatiotemporal control over drug availability could reduce or eliminate risks of systemic drug exposure, drug–drug interactions and toxicity.29–31 However, current implantable drug-eluting systems have a finite drug payload, inevitably requiring repeated administration of drugs or drug depots over time. For example, a single dose, tacrolimus-loaded gel injected into the limb transplants could prolong transplant survival for >100 days in rodent and porcine models, but eventually was depleted of drugs which led to graft rejection.32,33 Albeit the great promise and clinical translatable potential of local drug depots, there is currently no noninvasive technique to refill these depots once their payload is depleted. Surgical or invasive replacement or repeated administration is typically needed, but could cause graft inflammation and rejection. To address limitations of current approaches, we developed, for the first time, clickable and degradable immunosuppressive prodrugs that enable timely and repeated conjugation to biomaterials or implants via efficient Click chemistry, and allow for minimally invasive loading of depots previously placed in a tissue. We have previously demonstrated the feasibility of targeting and conjugating chemotherapeutic prodrugs to a hydrogel depot, in the context of cancer treatment.34,35
In the current study, prodrugs were designed to contain a clickable head, a water-soluble segment, and a degradable linker responsive to either pH, redox, enzymes or other triggers indicative of tissue rejection. The release of the water-soluble prodrug could then be autonomously increased from a baseline level if tissue rejection, as indicated by the level of the trigger agent (e.g., lower pH at the site of inflammation), is initiated. Both tacrolimus and rapamycin contain ketone groups for coupling of hydrazides and hydroxyl groups for esterification. Considering the much milder reaction conditions and the resulting acid-labile hydrazone bonds,36,37 hydrazide-ketone chemistry was utilized for synthesizing prodrugs of tacrolimus and rapamycin. On the other hand, MPA contains a hydroxyl group that can be converted to carbonates for the development of acid-labile prodrugs. For the clickable head, dibenzocyclooctyne (DBCO) was introduced to all drugs as it can react with azido groups via efficient Click chemistry. The copper-free Click chemistry between azide and DBCO has been widely used for in vivo applications, due to the high reaction efficiency and bioorthogonality under physiological conditions, small size of azides, and high stability of both azides and DBCO.38,39 Polyethylene glycol (PEG) was used as the water-solubilizing segment to enable pro-drugs to circulate in the body, diffuse into tissues, and bind to the hydrogel depot. To our knowledge, this is the first report of synthesis and characterization of DBCO-bearing prodrugs of tacrolimus, rapamycin and MPA, and their pH-dependent release profiles, pharmacokinetics, and Click chemistry-mediated targeting to azido-modified alginate hydrogels in vitro and after in vivo tissue implantation.
Results and discussion
We first modified rapamycin with clickable DBCO via a cleavable linker, while improving the drug water solubility. Rapamycin contains three hydroxyl groups that allow for esterification and two ketone groups that enable conjugation of hydrazides (Fig. 1a). Esterification requires a potent nucleophilic catalyst or high temperature, making site-specific modification of hydroxyl groups difficult. In contrast, the coupling reaction between ketones and hydrazides with weak acids as the catalyst can be efficient under much milder conditions. Rapamycin was first conjugated with N-β-maleimidopropionic acid hydrazide (BMPH) to yield rapamycin-mal with a maleimide ending group (Fig. 1a), which was confirmed by high performance liquid chromatography (HPLC) and mass spectrometry (MS) (Fig. S1†). DBCO-PEG1k-SH was obtained via the amine-carboxyl coupling reaction between DBCO-NHS and NH2-PEG1k-SH (Fig. 1a and S2†), which further reacted with rapamycin-mal to obtain DBCO-PEG1k-hydrazone-rapamycin (DBCO-hz-rapa, Fig. 1a). This reaction was validated via HPLC (Fig. 1b), and the structure of DBCO-hz-rapa was characterized by mass spectrum (Fig. 1c) and 1H NMR spectrum (Fig. 1d). DBCO-hz-rapa showed excellent water-solubility (>100 mg mL−1), and can be hydrolyzed into rapamycin in a pH-dependent manner, with faster reaction kinetics under pH 5.5 in comparison to pH 7.4 (Fig. 1e). To confirm its clickable property, DBCO-hz-rapa was incubated with azido-functionalized alginate (alginate-N3, Fig. S3†). HPLC confirmed the complete consumption of DBCO-hz-rapa within 10 min (Fig. 1f), substantiating efficient Click chemistry-mediated conjugation. It is noteworthy that roughly 100 azido groups were conjugated to each alginate strand for the alginate-N3 used in this study.40 | ||
| Fig. 1 Synthesis and characterizations of Clickable, acid labile DBCO-hz-rapa. (a) Synthetic route of DBCO-hz-rapa. (b) HPLC profiles of rapamycin-mal, DBCO-PEG1k-SH, and DBCO-hz-rapa, respectively. (c) Mass spectrum of DBCO-hz-rapa. The interval of 14.7 (m/z) indicates the existence of [M + 3H]3+. (d) 1H NMR spectrum of DBCO-hz-rapa. (e) pH-Dependent degradation profiles of DBCO-hz-rapa to rapamycin under pH 7.4 and 5.5, respectively. Data were presented as mean ± SD (n = 4), and analyzed by two-tailed t-test (0.01 < *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001). (f) HPLC traces of the mixture of DBCO-hz-rapa and alginate-N3, which showed rapid conjugation via Click chemistry. | ||
Similarly, tacrolimus bearing a ketone group was first conjugated with BMPH to yield tacrolimus-mal (Fig. 2a and S4†), which further coupled with DBCO-PEG1k-SH to yield DBCO-PEG1k-hydrazone-tacrolimus (DBCO-hz-tacro) (Fig. 2a). The reaction was confirmed via HPLC (Fig. 2b), and the chemical structure of DBCO-hz-tacro was well characterized by mass spectrum (Fig. 2c) and 1H NMR spectrum (Fig. 2d). DBCO-hz-tacro also showed excellent water solubility (>100 mg mL−1), and can be degraded into tacrolimus in a pH-dependent manner, with a faster release of tacrolimus under pH 5.5 in comparison to pH 7.4 (Fig. 2e). The ability of DBCO-hz-tacro to covalently conjugate to alginate-N3via efficient Click chemistry was also validated by HPLC (Fig. 2f).
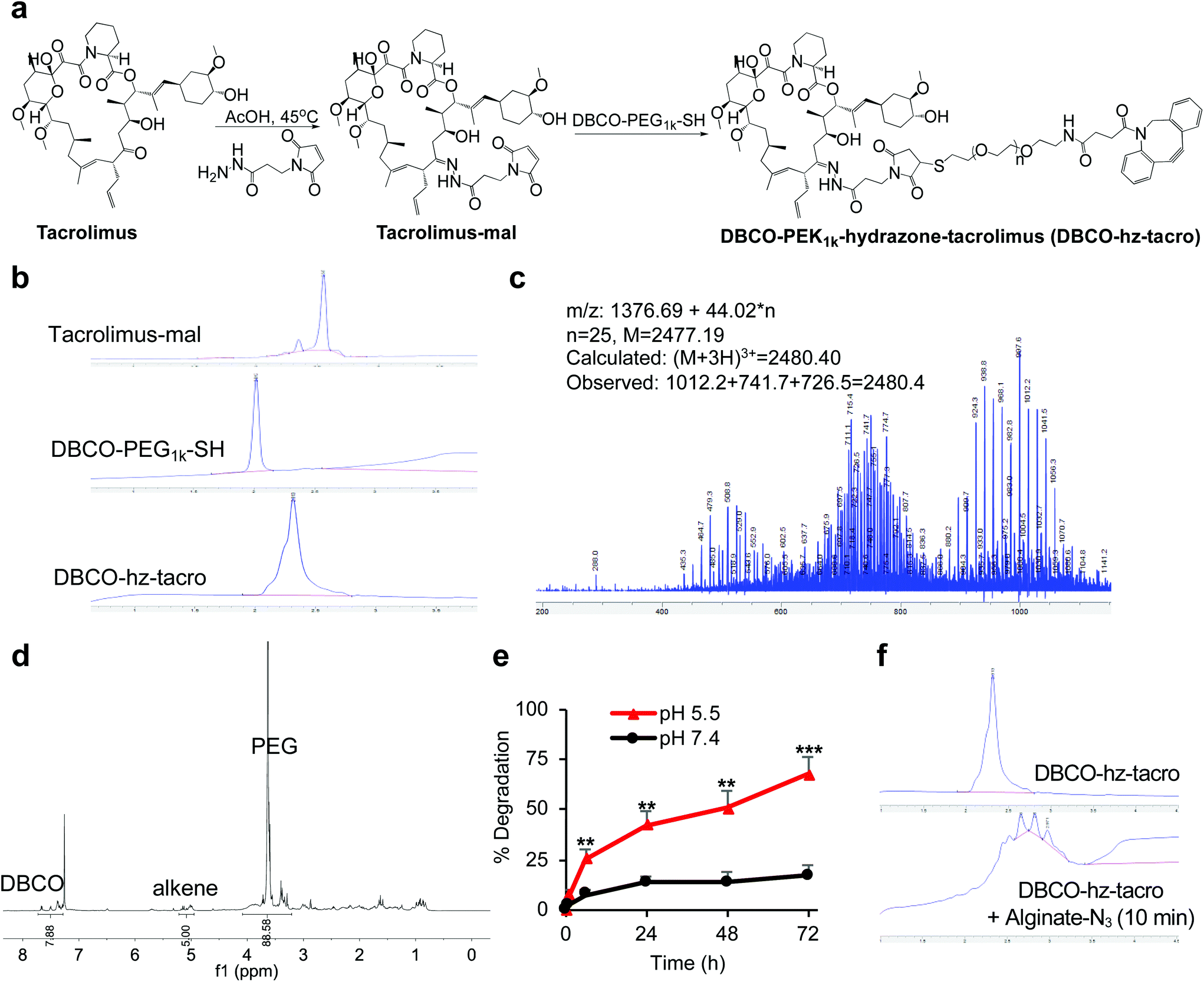 | ||
| Fig. 2 Synthesis and characterizations of clickable, acid labile DBCO-hz-tacro. (a) Synthetic route of DBCO-hz-tacro. (b) HPLC profiles of tacrolimus-mal, DBCO-PEG1k-SH, and DBCO-hz-tacro, respectively. (c) Mass spectrum of DBCO-hz-tacro. The interval of 14.7 (m/z) indicates the existence of [M + 3H]3+. (d) 1H NMR spectrum of DBCO-hz-tacro. (e) pH-Dependent degradation profiles of DBCO-hz-tacro to tacrolimus under pH 7.4 and 5.5, respectively. Data were presented as mean ± SD (n = 4), and analyzed by two-tailed t-test (0.01 < *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001). (f) HPLC traces of the mixture of DBCO-hz-tacro and alginate-N3, which showed rapid conjugation via Click chemistry. | ||
In contrast to rapamycin and tacrolimus, MPA only contains a hydroxyl group and a carboxyl group for functionalization (Fig. 3a). To avoid self-crosslinking, we converted the hydroxyl group into a carbonate by reacting with 4-nitrophenyl chloroformate, as confirmed by HPLC and mass spectrum (Fig. S5†). In parallel, DBCO-PEG1k-OH was synthesized via coupling of NH2-PEG1k-OH and DBCO-NHS (Fig. 3a and b). The nucleophilic attack of MPA-4-nitrophyenyl carbonate by DBCO-PEG1k-OH successfully yields DBCO-PEG1k-carbonate-MPA (DBCO-MPA) (Fig. 3b), as confirmed by mass spectra (Fig. 3c). DBCO-MPA with a high water-solubility (>250 mg mL−1) could be hydrolyzed to MPA in a pH-dependent manner (Fig. 3d). Similar to DBCO-hz-rapa and DBCO-hz-tacro, DBCO-MPA covalently conjugated to alginate-N3via Click chemistry (Fig. 3e).
 | ||
| Fig. 3 Synthesis and characterizations of clickable, acid labile DBCO-MPA. (a) Synthetic route of DBCO-MPA. (b) HPLC profiles of NH2-PEG1k-OH, DBCO-PEG1k-OH, and DBCO-MPA, respectively. (c) Mass spectra of NH2-PEG1k-OH, DBCO-PEG1k-OH, and DBCO-MPA, respectively. The interval of 22 (m/z) indicates the existence of [M + 2H]2+. (d) pH-Dependent degradation profiles of DBCO-MPA to MPA under pH 7.4 and 5.5, respectively. Data were presented as mean ± SD (n = 4), and analyzed by two-tailed t-test (0.01 < *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001). (e) HPLC traces of the mixture of DBCO-MPA and alginate-N3, which showed rapid conjugation via Click chemistry. | ||
After developing Clickable, acid-labile DBCO-hz-rapa, DBCO-hz-tacro, and DBCO-MPA, and demonstrated their capability to covalently conjugate to alginate-N3 polymer, we next studied whether alginate-N3 gels, formed by crosslinking of alginate-N3 polymer chains via calcium ions, can first capture these prodrugs and subsequently release the immunosuppressive drugs in a controlled manner (Fig. 4a). Alginate-N3 gels were prepared by mixing a 2% (w/v) alginate-N3 solution and a slurry of calcium sulfate for 45 min, and casting into cylindrical discs. Unmodified alginate gels were used as controls. After incubating gels with an aqueous solution of DBCO-hz-rapa, residual DBCO-hz-rapa in the solution was quantified via LC-MS. Alginate-N3 gels captured 26% of the DBCO-hz-rapa in solution within 5 minutes, which compared to less than 2% captured by control gels (Fig. 4b). At 30 min, 35% and 2% of solution DBCO-hz-rapa was captured by alginate-N3 and control alginate gels, respectively (Fig. 4b). The loading of DBCO-hz-rapa approached saturation after 3 h in both alginate-N3 and control alginate gels, and alginate-N3 gels exhibited ∼2-fold greater loading than control gels (Fig. S6†). The loading of control gels with DBCO-hz-rapa is expected to result from simple diffusion of the molecule into the hydrogel from the surrounding solution, which increased over time as expected. In comparison, water-insoluble rapamycin showed minimal diffusion into alginate-N3 gels (Fig. S7†). Following the loading study, the release kinetics of rapamycin from DBCO-hz-rapa conjugated alginate-N3 gels was characterized under different pH conditions. Alginate-N3 gels slowly released rapamycin over a course of >40 days, with faster release kinetics under pH 5.5, in comparison to pH 7.4 (Fig. 4c), presumably because of pH-dependent cleavage of the hydrazone linker of DBCO-hz-rapa. In contrast, control alginate gels exhibited fast release of the diffusively loaded DBCO-hz-rapa under neutral conditions, with over 70% being released within 72 h (Fig. S8†), demonstrating the importance of covalent conjugation to achieve sustained release of these free drugs over a long period.
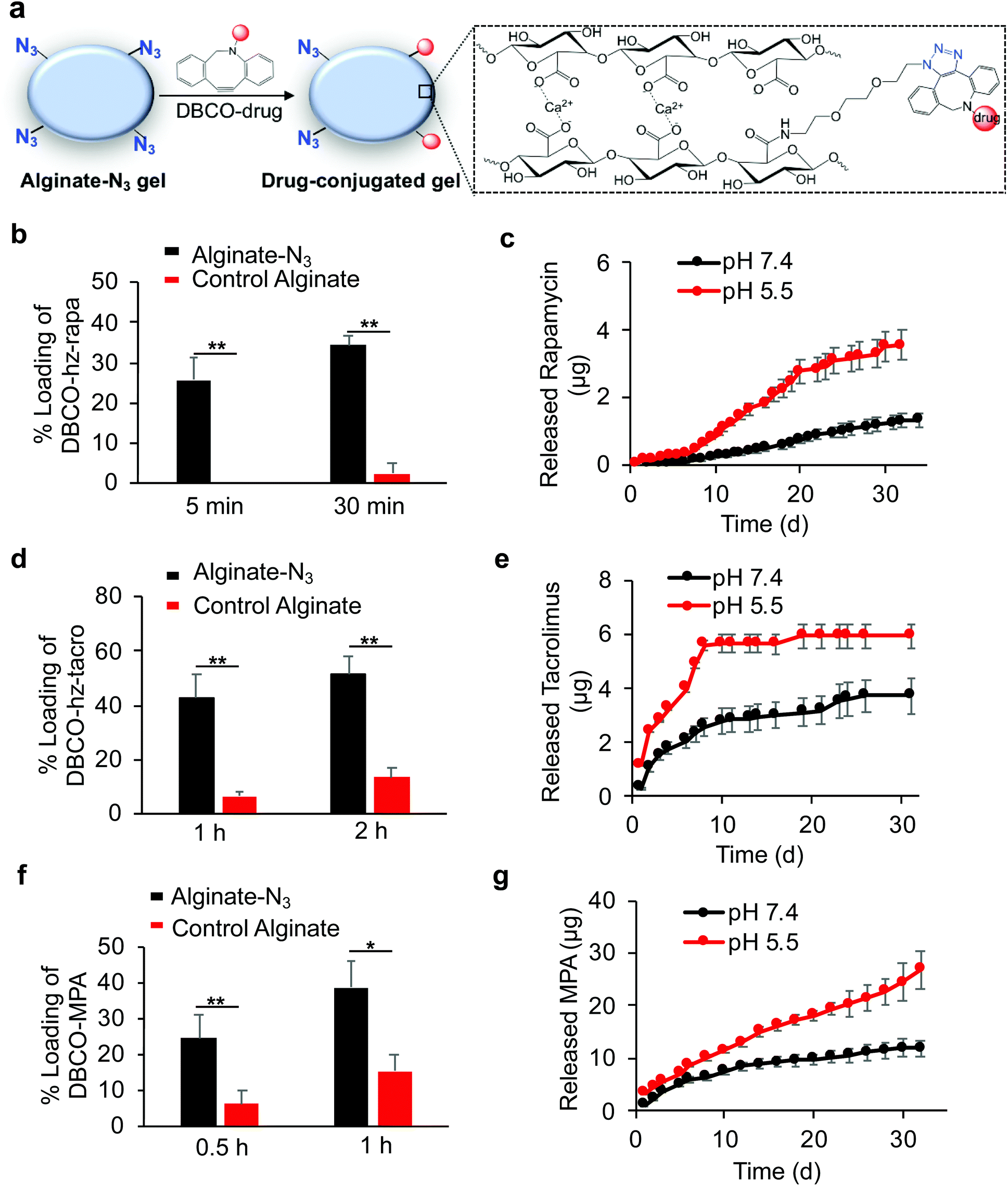 | ||
| Fig. 4 Targeted loading of DBCO-hz-rapa, DBCO-hz-tacro, and DBCO-MPA to alginate-N3 gels via Click chemistry and subsequent drug release kinetics. (a) Schematic illustration of coupling of DBCO-drugs with alginate-N3 gels. Gels (2 mm height, 4 mm diameter) in 24-well plates were incubated with prodrugs in DMEM at 37 °C. At different time points, 10 μL of medium was taken out to quantify unconjugated DBCO-drugs. After 24 h, gels were placed in fresh medium to monitor the release of active drugs. (b) Loading of DBCO-hz-rapa into alginate-N3 and unmodified alginate gels, respectively after 5 min or 30 min incubation (n = 4). (c) Release kinetics of rapamycin from alginate-N3 gels at pH 5.5 and pH 7.4, respectively (n = 4). (d) Loading of DBCO-hz-tacro into alginate-N3 and unmodified alginate gels, respectively after 1 h or 2 h incubation (n = 4). (e) Release kinetics of tacrolimus from alginate-N3 gels at pH 5.5 and pH 7.4, respectively (n = 4). (f) Loading of DBCO-MPA into alginate-N3 and unmodified alginate gels, respectively after 0.5 h or 1 h incubation (n = 4). (g) Release kinetics of MPA from alginate-N3 gels at pH 5.5 and pH 7.4, respectively (n = 4). Data presented as mean ± SD and analyzed by two-tailed t-test (0.01 < *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001). | ||
Similar to DBCO-hz-rapa, DBCO-hz-tacro was also rapidly captured by alginate-N3 gels via efficient Click chemistry, with 5.7-fold and 2.9-fold increases in loading compared to control alginate gels after 1 h and 2 h, respectively (Fig. 4d). The alginate-N3 gels captured 160% more DBCO-hz-tacro than control gels after 24 h (Fig. S9†). DBCO-hz-tacro conjugated alginate-N3 gels subsequently released tacrolimus in a pH-dependent manner, with faster release kinetics under pH 5.5 in comparison to pH 7.4 (Fig. 4e). DBCO-hz-tacro loaded alginate-N3 gels started with a faster rate of release, and reached the steady release phase at an earlier time, in comparison to the DBCO-hz-rapa (Fig. 4c and e).
DBCO-MPA was also successfully covalently captured by alginate-N3 gels via Click chemistry in vitro, with a 2.9-fold and 1.5-fold loading increase within 0.5 h and 1 h, respectively in comparison to control alginate gels (Fig. 4f). Alginate-N3 gels still captured 145% more DBCO-MPA than control gels at 12 h (Fig. S10†). DBCO-MPA conjugated alginate-N3 gels slowly released MPA over a course of >30 days, and the release of MPA was faster at pH 5.5 compared to pH 7.4 (Fig. 4g). Compared to DBCO-hz-rapa and DBCO-hz-tacro, DBCO-MPA loaded alginate-N3 gels showed a faster release of drugs in both acidic and neutral conditions (Fig. 4c, e, and f), presumably due to the significantly greater hydrophilicity of DBCO-MPA.
The pharmacokinetic properties of DBCO-hz-rapa, DBCO-hz-tacro, and DBCO-MPA were next examined to determine if their circulation times would be sufficiently long to allow for minimally invasive loading onto hydrogel depots, but would be short enough to minimize systemic availability of active drug before clearance of unbound conjugates. Balb/c mice were intravenously (i.v.) injected with prodrugs, and blood was collected for prodrug quantification at different time points. DBCO-hz-rapa was rapidly cleared from the blood, with a blood concentration of 28 μg mL−1 (∼7% of injected dose (I.D.)) at 1 h post injection (Fig. S11†). The majority of injected DBCO-hz-rapa was cleared from the bloodstream within 9 h (Fig. S11†). For DBCO-hz-tacro, ∼11% was retained in the blood after 12 min, and the level dropped to ∼1.2% I.D. at 6 h (Fig. S12†). Compared to DBCO-hz-rapa and DBCO-hz-tacro, DBCO-MPA showed a slightly longer retention in the bloodstream, with 7.6% I.D. at 2 h and 3.1% I.D. at 6 h (Fig. S13†), presumably due to the better water-solubility of DBCO-MPA. Compared to DBCO-MPA, free MPA became undetectable within 2 h post intravenous injection, which is consistent with previous reports in rats.41 For all three prodrugs, the vast majority was cleared from the bloodstream by 12 h (Fig. S11–13†). In clinic, free rapamycin, tacrolimus, and MPA are often orally administered in formulations containing oils or as tablets.42–44 However, orally administered free drugs suffer from significant gastrointestinal degradation, low total bioavailability, variable pharmacokinetics, and dominant existence of drug–protein complexes in the bloodstream;42–47 these issues can be potentially addressed by the use of intravenously administered, water-soluble, and anti-fouling prodrugs. Compared to reported drug-encapsulating nanoparticles,40,48,49 the blood circulation time of DBCO-hz-rapa, DBCO-hz-tacro, and DBCO-MPA is comparable or shorter, which is important to minimize the hydrolysis window of prodrugs and thus the systemic exposure of released free drugs.
We next studied whether alginate-N3 gels can efficiently capture DBCO-modified immunosuppressive drugs in vivo. In the first studies, Balb/c mice were subcutaneously injected with alginate-N3 or control alginate gels, followed by i.v. injection of DBCO-hz-rapa 1 h later. At 48 h post injection of DBCO-hz-rapa, gels were harvested, disassociated, and treated with acids, prior to quantification of recovered rapamycin via LC-MS (Fig. S14a†). Compared to control gels, alginate-N3 gels retained a significantly higher amount of rapamycin (Fig. S14b and c†), indicating the enhanced capture of DBCO-hz-rapa by alginate-N3 gels via Click chemistry. To further confirm this targeting effect, alginate-N3 gels and control gels were subcutaneously injected into the left and right flanks of the same Balb/c mice, and after 1 h, DBCO-hz-rapa was i.v. injected via tail vein (Fig. 5a). At 48 h post injection of DBCO-hz-rapa, retained rapamycin was quantified and compared. Alginate-N3 gels were able to capture significantly more DBCO-hz-rapa, as compared to control gels, with a 130% increase in rapamycin retention (Fig. 5b). Approximately 1% of I.D. of DBCO-hz-rapa was retained in alginate-N3 gels at 48 h post injection, which was significantly higher than control gels (Fig. 5c). Similar studies involving i.v. infusion of DBCO-hz-tacro and DBCO-MPA into Balb/c mice bearing alginate-N3 and control gels confirmed that both tacrolimus and MPA pro-drugs could be captured by alginate-N3 gels in vivo (Fig. 5d and e). Altogether, these experiments demonstrate that alginate-N3 gels can covalently capture DBCO-functionalized immunosuppressive drugs via efficient Click chemistry in vivo. Past studies have demonstrated that click chemistry-mediated targeting efficiency varies with the types of molecules and locations of tissues or biomaterials which are targeted. Molecules that readily diffuse into and are rapidly cleared from target tissues or biomaterials exhibit high targeting efficiency (>100% improvement over non-targeted molecules),34,39,49 while molecules with a bulky chemical structure and relatively poor pharmacokinetics have shown limited targeting effect (0%–50% improvement).50–52 The effective targeting of DBCO-hz-rapa (130% improvement), DBCO-hz-tacro (105% improvement) and DBCO-MPA (80% improvement) likely results from their good pharmacokinetics and rapid clearance of uncoupled drug from gels.
 | ||
| Fig. 5 Targeted loading of DBCO-hz-rapa, DBCO-hz-tacro, and DBCO-MPA to alginate-N3 gels in vivo via click chemistry. (a) Schematic illustration of in vivo loading study using mice bearing alginate-N3 and control alginate gels on different flanks. DBCO-hz-rapa (150 mg kg−1) or DBCO-hz-tacro (150 mg kg−1) or DBCO-MPA (100 mg kg−1) were i.v. injected via tail vein, and gels subsequently retrieved to quantify bound prodrugs. (b) Amount of rapamycin recovered from alginate-N3 or control alginate gels at 48 h post injection (n = 4). (c) Amount of loaded rapamycin in the form of percentage of injected dose (n = 4). (d) Amount of tacrolimus recovered from alginate-N3 or control alginate gels at 48 h post injection (n = 4). (e) Amount of MPA recovered from alginate-N3 or control alginate gels at 48 h post injection (n = 4). Data presented as mean ± SD and analyzed by two-tailed t-test (0.01 < *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001). | ||
Conclusion
We have developed clickable, acid-labile DBCO-hz-tacro, DBCO-hz-rapa and DBCO-MPA for in vivo targeted delivery and subsequent controlled, local release of immunosuppressive drugs. These prodrugs enabled covalent conjugation to azido-modified alginate gels, resulting in a sustained, pH-dependent release of active drugs in vitro. We further demonstrated that subcutaneously injected alginate-N3 gels can covalently capture i.v. injected DBCO-hz-tacro, DBCO-hz-rapa and DBCO-MPA. This approach potentially enables targeted delivery of immunosuppressive drugs to biomaterial depots at any time of interest, followed by sustained release of active drugs locally, which is anticipated to flexibly maintain immunosuppression after allotransplantation. Alginate gels as evaluated in our platform are natural non-immunogenic polysaccharides, enable controlled local release of drugs, and are also refillable by binding fresh intravenously injected prodrugs. These gels, once implanted in tissues, do not require replacement every time the drug levels fall to sub-therapeutic thresholds, thus avoiding trauma and inflammation due to repeated gel injections. Further evaluation of the therapeutic benefits of these prodrugs in the context of reconstructive transplantation is underway in animal models.Methods
Materials and instrumentation
N-β-Maleimidopropionic acid hydrazide (BMPH) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Rapamycin and tacrolimus were purchased from LC Laboratories (Woburn, MA, USA). MPA was purchased from Life Technologies Corporation (Chicago, IL, USA). NH2-PEG1k-SH and NH2-PEG1k-OH were purchased from Laysan Bio (Arab, AL, USA). DBCO-NHS was purchased from Conju Probe (San Diego, CA, USA). 11-Azido-3,6,9-trioxaundecan-1-amine and other compounds were purchased from Sigma Aldrich (St Louis, MO, USA) unless otherwise noted. PRONOVA UP MVG sodium alginate (endotoxin-free) was purchased from Fmc Biopolymer AS (Sandvika, Norway). Small compounds were run on the Agilent 1290/6140 ultra high performance liquid chromatography/mass spectrometer (LC-MS). Compounds were purified using the Agilent 1200 preparative high performance liquid chromatography (HPLC). Proton nuclear magnetic resonance spectra were collected on the Agilent DD2 600.Animals
BALB/c mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Feed and water were available ad libitum. Artificial light was provided in a 12 h/12 h cycle. All procedures involving animals were done in compliance with National Institutes of Health and Institutional guidelines with approval of Harvard University's Institutional Animal Care and Use Committee.Synthesis of rapamycin-mal
Rapamycin (0.1 mmol) and BMPH (0.12 mmol) were dissolved in methanol, followed by the addition of glacial acetic acid (5 μL). The mixture was stirred at 45 °C for 48 h. After removal of the solvent, the crude product was purified by preparative HPLC to yield rapamycin-mal (60% yield). MS m/z: calculated for C58H87N4O15 [M + H]+ 1079.6, found 1079.4, calculated for C58H85N4O15 [M − H]− 1077.6, found 1077.3.Synthesis of DBCO-PEG1k-SH
NH2-PEG1k-SH (0.1 mmol) and DBCO-NHS (0.1 mmol) were dissolved in methanol. The mixture was stirred at 37 °C for 24 h. After removal of the solvent, the crude product was stored for use.Synthesis of DBCO-hz-rapa
Rapamycin-mal (0.1 mmol) and DBCO-PEG1k-SH (0.1 mmol) were dissolved in methanol, followed by the addition of triethylamine (0.1 mmol). The mixture was stirred at 40 °C for 48 h. After removal of the solvent, the crude product was purified via preparative HPLC to yield DBCO-hz-rapa.Synthesis of tacrolimus-mal
Tacrolimus (0.1 mmol) and BMPH (0.12 mmol) were dissolved in methanol, followed by the addition of glacial acetic acid (5 μL). The mixture was stirred at 45 °C for 48 h. After removal of the solvent, the crude product was purified by preparative HPLC to yield tacrolimus-mal (70% yield). MS m/z: calculated for C52H78N3O13 [M + H]+ 969.5, found 969.4.Synthesis of DBCO-hz-tacro
Tacrolimus-mal (0.1 mmol) and DBCO-PEG1k-SH (0.1 mmol) were dissolved in methanol, followed by the addition of triethylamine (0.1 mmol). The mixture was stirred at 40 °C for 48 h. After removal of the solvent, the crude product was purified via preparative HPLC to yield DBCO-hz-tacro.Synthesis of MPA-4-nitrophenylcarbonate
MPA (0.1 mmol) and 4-nitrophenyl chloroformate (0.11 mmol) were dissolved in anhydrous dichloromethane, followed by the addition of triethylamine (0.1 mmol). The mixture was stirred at room temperature for 24 h. After removal of the solvent, the crude product was purified via preparative HPLC to yield MPA-4-nitrophenylcarbonate (75% yield). MS m/z: calculated for C24H24NO10 [M + H]+ 486.1, found 486.0, calculated for [2M + Na]+ 993.2, found 993.0.Synthesis of DBCO-PEG1k-OH
NH2-PEG1k-OH (0.1 mmol) and DBCO-NHS (0.1 mmol) were dissolved in methanol. The mixture was stirred at 37 °C for 24 h. After removal of the solvent, the crude product was purified via preparative HPLC.Synthesis of DBCO-MPA
MPA-4-nitrophenylcarbonate (0.1 mmol) and DBCO-PEG1k-OH (0.1 mmol) were dissolved in dimethylformamide, followed by addition of N,N-diisopropylethylamine (0.1 mmol) and 4-dimethylaminopyridine (0.1 mmol). The mixture was stirred at room temperature for 48 h. After removal of the solvent, the crude product was purified via preparative HPLC to yield DBCO-MPA.Synthesis of alginate-N3
Alginate-N3 was synthesized following the reported procedure.40 Alginate (1 g) was dissolved in MES buffer, followed by the addition of 11-azido-3,6,9-trioxaundecan-1-amine (160 mg). The mixture was stirred at room temperature for 30 min, and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl, 160 mg) and hydroxybenzotriazole (HOBt, 140 mg) were then added. The reaction mixture was stirred for another 48 h, adjusted to pH 7.4 using sodium hydroxide solution, placed in a dialysis bag (3.5k MWCO), dialyzed against DI water for 3 days (medium change every 12 h), lyophilized, and stored for use.Preparation of alginate-N3 gels
2% (w/v) alginate-N3 in Dulbecco's Modified Eagle Medium (DMEM, 3 mL) was added into the first syringe, and calcium sulfate slurry (150 μL) was added into the second prepared syringe. Air bubbles were removed, and the syringes were connected by a luer-lock. Solutions were mixed for 20–30 times by pushing plungers back and forth very rapidly. The calcium sulfate slurry was evenly mixed throughout the syringe. For in vitro studies, the gels were immediately cast between two silanized glass plates separated by 2 mm spacers. After allowing the gels to cross-link for 45 min, gel disks were punched out using a sterile biopsy punch. For in vivo studies, gel solutions (100 μL) were freshly prepared and subcutaneously injected via an 18 G needle.Loading of DBCO-functionalized drugs to alginate-N3 gels in vitro
Alginate-N3 gels or unmodified alginate gels (2 mm height, 4 mm diameter) in 24-well plates were incubated with DBCO-hz-rapa, DBCO-hz-tacro or DBCO-MPA in DMEM (300 μL, 10 mg mL−1) at 37 °C. At different time points, 10 μL of medium was taken out and diluted with 90 μL of acetonitrile prior to LC-MS analyses. The percentage of prodrug loading was calculated as: (amount of fed prodrugs − amount of remaining prodrugs)/(amount of fed prodrugs). For diffusion study of rapamycin, rapamycin was dissolved in dimethyl sulfoxide prior to addition into the medium.Release study of drug-loaded gels
Alginate-N3 gels or control alginate gels previously incubated with DBCO-hz-rapa, DBCO-hz-tacro or DBCO-MPA for 24 h were incubated at 37 °C in fresh DMEM with a pH value of 7.4 and 5.5, respectively. At different time points, 10 μL of medium was taken out and diluted with 90 μL of acetonitrile prior to LC-MS analyses.Pharmacokinetics study of DBCO-functionalized drugs
DBCO-hz-rapa (50 mg kg−1), DBCO-hz-tacro (50 mg kg−1) or DBCO-MPA (50 mg kg−1) were i.v. administered to Balb/c mice. Blood was collected at different times post injection of DBCO-functionalized drugs, treated with lysis buffer, and centrifuged. Supernatants were collected for quantification of prodrugs via LC-MS.In vivo targeting of DBCO-functionalized drugs
Balb/c mice were subcutaneously injected with alginate-N3 gels or control gels (100 μL). After 1 h, DBCO-hz-rapa (150 mg kg−1), DBCO-hz-tacro (150 mg kg−1) or DBCO-MPA (100 mg kg−1) were intravenously injected via tail vein. At 48 h post injection of DBCO-functionalized drugs, gels were harvested, disrupted using EDTA (10 mM) on ice, and treated with formic acid/acetonitrile to release free drugs. After centrifugation, supernatants were analyzed by LC-MS to quantify the retained drug.Statistical analyses
Statistical analysis was performed using GraphPad Prism and Microsoft Excel. Sample variance was tested using the F test. For samples with equal variance, the significance between the groups was analyzed by a two-tailed student's t test. For samples with unequal variance, a two-tailed Welch's t-test was performed. The results were deemed significant at 0.01 < *P ≤ 0.05, highly significant at 0.001 < **P ≤ 0.01, and extremely significant at ***P ≤ 0.001.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The studies were supported by funding from the Department of Defense (W81XWH-15-2-0017, Log MR141089) and from National Institutes of Health (1 R01 EB023287, 1 R01 CA223255). H. W. gratefully acknowledges funding support from the Wyss Technology Development Fellowship.References
- C. L. Kaufman, et al., Current Status of Vascularized Composite Allotransplantation, Am. Surg., 2019, 85, 631–637 Search PubMed.
- J. T. Shores, G. Brandacher and W. P. Lee, Hand and upper extremity transplantation: an update of outcomes in the worldwide experience, Plast. Reconstr. Surg., 2015, 135, 351e–360e CrossRef CAS.
- V. S. Gorantla, J. A. Plock and M. R. Davis, Anesthesia and Perioperative Care for Organ Transplantation, Springer, New York, 2016, pp. 539–552 Search PubMed.
- C. F. Barker and J. F. Markmann, Historical overview of transplantation, Cold Spring Harbor Perspect. Med., 2013, 3, a014977 CrossRef PubMed.
- V. S. Gorantla, et al., Immunosuppressive agents in transplantation: mechanisms of action and current anti-rejection strategies, Microsurgery, 2000, 20, 420–429 CrossRef CAS.
- A. Thomson, C. Bonham and A. Zeevi, Mode of action of tacrolimus (FK506): molecular and cellular mechanisms, Ther. Drug Monit., 1995, 17, 584–591 CrossRef CAS.
- T. R. Brazelton, Molecular mechanisms of action of new xenobiotic immunosuppressive drugs: tacrolimus (FK506), sirolimus (rapamycin), mycophenolate mofetil and leflunomide, Curr. Opin. Immunol., 1996, 8, 710–720 CrossRef CAS.
- F. J. Dumont and Q. Su, Mechanism of action of the immunosuppressant rapamycin, Life Sci., 1995, 58, 373–395 CrossRef CAS.
- S. N. Sehgal, Rapamune®(RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression, Clin. Biochem., 1998, 31, 335–340 CrossRef CAS.
- M. Poon, et al., Rapamycin inhibits vascular smooth muscle cell migration, J. Clin. Invest., 1996, 98, 2277–2283 CrossRef CAS.
- J. E. S. Kitchin, M. K. Pomeranz, G. Pak, K. Washenik and J. L. Shupack, Rediscovering mycophenolic acid: a review of its mechanism, side effects, and potential uses, J. Am. Acad. Dermatol., 1997, 37, 445–449 CrossRef CAS.
- A. C. Allison, W. J. Kowalski, C. D. Muller and E. M. Eugui, Mechanisms of action of mycophenolic acid, Ann. N. Y. Acad. Sci., 1993, 696, 63–87 CrossRef CAS.
- A. C. Allison and E. M. Eugui, Mechanisms of action of mycophenolate mofetil in preventing acute and chronic allograft rejection, Transplantation, 2005, 80, S181–S190 CrossRef CAS.
- R. Bentley, Mycophenolic Acid: A One Hundred Year Odyssey from Antibiotic to Immunosuppressant, Chem. Rev., 2000, 100, 3801–3826 CrossRef CAS.
- W. Land and F. Vincenti, Toxicity-Sparing Protocols Using Mycophenolate Mofetil in Renal Transplantation, Transplantation, 2005, 80, S221–S234 CrossRef CAS.
- S. N. Sehgal, Rapamune (Sirolimus, rapamycin): an overview and mechanism of action, Ther. Drug Monit., 1995, 17, 660–665 CrossRef CAS PubMed.
- S. F. Stewart, M. Hudson, D. Talbot, D. Manas and C. P. Day, Mycophenolate mofetil monotherapy in liver transplantation, Lancet, 2001, 357, 609–610 CrossRef CAS.
- V. S. Gorantla and A. J. Demetris, Acute and chronic rejection in upper extremity transplantation: what have we learned?, Hand Clin., 2011, 27, 481–493 CrossRef.
- E. Brooks, S. E. Tett, N. M. Isbel and C. E. Staatz, Population Pharmacokinetic Modelling and Bayesian Estimation of Tacrolimus Exposure: Is this Clinically Useful for Dosage Prediction Yet?, Clin. Pharmacokinet., 2016, 55, 1295–1335 CrossRef CAS.
- R. Bouamar, et al., Tacrolimus predose concentrations do not predict the risk of acute rejection after renal transplantation: a pooled analysis from three randomized-controlled clinical trials(dagger), Am. J. Transplant., 2013, 13, 1253–1261 CrossRef CAS PubMed.
- G. Brandacher, W. P. Lee and S. Schneeberger, Minimizing immunosuppression in hand transplantation, Expert Rev. Clin. Immunol., 2012, 8, 673–683 CrossRef CAS.
- J. T. Schnider, et al., Site-specific immunosuppression in vascularized composite allotransplantation: prospects and potential, Clin. Dev. Immunol., 2013, 2013, 495212 Search PubMed.
- J. Sellares, et al., Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence, Am. J. Transplant., 2012, 12, 388–399 CrossRef CAS.
- G. Janka, S. Imashuku, G. Elinder, M. Schneider and J.-I. Henter, Infection-and malignancy-associated hemophagocytic syndromes: secondary hemophagocytic lymphohistiocytosis, Hematol. Oncol. Clin., 1998, 12, 435–444 CrossRef CAS.
- R. T. Bustami, et al., Immunosuppression and the risk of post-transplant malignancy among cadaveric first kidney transplant recipients, Am. J. Transplant., 2004, 4, 87–93 CrossRef.
- A. Gutierrez-Dalmau and J. M. Campistol, Immunosuppressive therapy and malignancy in organ transplant recipients, Drugs, 2007, 67, 1167–1198 CrossRef CAS PubMed.
- A. Capron, et al., Correlation of tacrolimus levels in peripheral blood mononuclear cells with histological staging of rejection after liver transplantation: preliminary results of a prospective study, Transplant Int., 2012, 25, 41–47 CrossRef CAS.
- F. G. Feturi, et al., Mycophenolic Acid for Topical Immunosuppression in Vascularized Composite Allotransplantation: Optimizing Formulation and Preliminary Evaluation of Bioavailability and Pharmacokinetics, Front. Surg., 2018, 5, 20 CrossRef.
- J. T. Schnider, et al., Site-specific immunosuppression in vascularized composite allotransplantation: prospects and potential, Clin. Dev. Immunol., 2013, 2013, 495212 Search PubMed.
- T. Ruers, W. Buurman, J. Smits, H. Struyker-Boudier and G. Kootstra, Local treatment of renal allografts, a promising way to reduce the dosage of immunosuppressive drugs. Comparison of various ways of administering prednisolone, Transplantation, 1986, 41, 156–161 CrossRef CAS.
- V. Holan, et al., Cyclosporine A-loaded and stem cell-seeded electrospun nanofibers for cell-based therapy and local immunosuppression, J. Controlled Release, 2011, 156, 406–412 CrossRef CAS.
- T. Gajanayake, et al., A single localized dose of enzyme-responsive hydrogel improves long-term survival of a vascularized composite allograft, Sci. Transl. Med., 2014, 6, 249ra110 CrossRef.
- C. A. Fries, et al., Graft-implanted, enzyme responsive, tacrolimus-eluting hydrogel enables long-term survival of orthotopic porcine limb vascularized composite allografts: A proof of concept study, PLoS One, 2019, 14, e0210914 CrossRef CAS.
- Y. Brudno, et al., Replenishable drug depot to combat post-resection cancer recurrence, Biomaterials, 2018, 178, 373–382 CrossRef CAS.
- Y. Brudno, et al., Refilling drug delivery depots through the blood, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12722–12727 CrossRef CAS.
- E. T. Kool, D.-H. Park and P. Crisalli, Fast hydrazone reactants: electronic and acid/base effects strongly influence rate at biological pH, J. Am. Chem. Soc., 2013, 135, 17663–17666 CrossRef CAS PubMed.
- S. J. Sonawane, R. S. Kalhapure and T. Govender, Hydrazone linkages in pH responsive drug delivery systems, Eur. J. Pharm. Sci., 2017, 99, 45–65 CrossRef CAS.
- J. M. Baskin, et al., Copper-free click chemistry for dynamic in vivo imaging, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS.
- H. Wang, et al., Selective in vivo metabolic cell-labeling-mediated cancer targeting, Nat. Chem. Biol., 2017, 13, 415 CrossRef CAS.
- Y. Brudno, et al., In Vivo Targeting through Click Chemistry, ChemMedChem, 2015, 10, 617–620 CrossRef CAS.
- H. Tian, J. Ou, S. C. Strom and R. Venkataramanan, Pharmacokinetics of tacrolimus and mycophenolic acid are altered, but recovered at different times during hepatic regeneration in rats, Drug Metab. Dispos., 2005, 33, 329–335 CrossRef CAS.
- K. Mahalati and B. D. Kahan, Clinical Pharmacokinetics of Sirolimus, Clin. Pharmacokinet., 2001, 40, 573–585 CrossRef CAS PubMed.
- D. Zhang and D. S.-L. Chow, Clinical Pharmacokinetics of Mycophenolic Acid in Hematopoietic Stem Cell Transplantation Recipients, Eur. J. Drug Metab. Pharmacokinet., 2017, 42, 183–189 CrossRef CAS.
- R. Venkataramanan, et al., Clinical Pharmacokinetics of Tacrolimus, Clin. Pharmacokinet., 1995, 29, 404–430 CrossRef CAS.
- A. MacDonald, J. Scarola, J. T. Burke and J. J. Zimmerman, Clinical pharmacokinetics and therapeutic drug monitoring of sirolimus, Clin. Ther., 2000, 22, B101–B121 CrossRef CAS.
- M. Antignac, B. Barrou, R. Farinotti, P. Lechat and S. Urien, Population pharmacokinetics and bioavailability of tacrolimus in kidney transplant patients, Br. J. Clin. Pharmacol., 2007, 64, 750–757 CAS.
- T. Shirasu, et al., Nanoparticles Effectively Target Rapamycin Delivery to Sites of Experimental Aortic Aneurysm in Rats, PLoS One, 2016, 11, e0157813–e0157813 CrossRef.
- S.-B. Shin, H.-Y. Cho, D.-D. Kim, H.-G. Choi and Y.-B. Lee, Preparation and evaluation of tacrolimus-loaded nanoparticles for lymphatic delivery, Eur. J. Pharm. Biopharm., 2010, 74, 164–171 CrossRef CAS.
- T. Solymosi, et al., Sirolimus formulation with improved pharmacokinetic properties produced by a continuous flow method, Eur. J. Pharm. Biopharm., 2015, 94, 135–140 CrossRef CAS.
- H. Wang, et al., In vivo cancer targeting via glycopolyester nanoparticle mediated metabolic cell labeling followed by click reaction, Biomaterials, 2019, 119305 CrossRef CAS.
- H. Wang, et al., Targeted Ultrasound-Assisted Cancer-Selective Chemical Labeling and Subsequent Cancer Imaging using Click Chemistry, Angew. Chem., Int. Ed., 2016, 55, 5452–5456 CrossRef CAS.
- H. Wang, et al., In Vivo Targeting of Metabolically Labeled Cancers with Ultra-Small Silica Nanoconjugates, Theranostics, 2016, 6, 1467–1476 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ESI Fig. S1–S13. See DOI: 10.1039/c9bm01487j |
| This journal is © The Royal Society of Chemistry 2020 |