
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substituting HF by HBF4 – an optimized digestion method for multi-elemental sediment analysis via ICP-MS/MS†
Tristan
Zimmermann
 a,
Marcus
von der Au
b,
Anna
Reese
ac,
Ole
Klein
ac,
Lars
Hildebrandt
ac and
Daniel
Pröfrock
*a
a,
Marcus
von der Au
b,
Anna
Reese
ac,
Ole
Klein
ac,
Lars
Hildebrandt
ac and
Daniel
Pröfrock
*a
aHelmholtz-Zentrum Geesthacht, Institute of Coastal Research, Marine Bioanalytical Chemistry, Max-Planck Str. 1, 21502 Geesthacht, Germany. E-mail: daniel.proefrock@hzg.de
bBundesanstalt für Materialforschung und -prüfung, Fachbereich 1.1, Richard-Willstätter-Straße 11, 12489 Berlin, Germany
cDepartment of Chemistry, Inorganic and Applied Chemistry, Universität Hamburg, Martin-Luther-King-Platz 6, 20146 Hamburg, Germany
First published on 24th July 2020
Abstract
Determination of elemental mass fractions in sediments plays a major role in evaluating the environmental status of aquatic ecosystems. Herewith, the optimization of a new total digestion protocol and the subsequent analysis of 48 elements in different sediment reference materials (NIST SRM 2702, GBW 07313, GBW 07311 and JMC-2) based on ICP-MS/MS detection is presented. The developed method applies microwave acid digestion and utilizes HBF4 as fluoride source for silicate decomposition. Similar to established protocols based on HF, HBF4 ensures the dissolution of the silicate matrix, as well as other refractory oxides. As HBF4 is not acutely toxic; no special precautions have to be made and digests can be directly measured via ICP-MS without specific sample inlet systems, evaporation steps or the addition of e.g. H3BO3, in order to mask excess HF. Different acid mixtures with and without HBF4 were evaluated in terms of digestion efficiency based on the trace metal recovery. The optimized protocol (5 mL HNO3, 2 mL HCL, 1 mL HBF4) allows a complete dissolution of the analyzed reference materials, as well as quantitative recoveries for a wide variety of certified analytes. Low recoveries for e.g. Sr, Ba and rare earth elements due to fluoride precipitation of HF-based digestions protocols, can be avoided by the usage of HBF4 instead. Based on the usage of high purity HBF4 all relevant trace, as well as matrix elements can be analyzed with sufficiently low LOQs (0.002 μg L−1 for U up to 6.7 μg L−1 for Al). In total, 34 elements were within a recovery range of 80%–120% for all three analyzed reference materials GBW 07313, GBW 07311 and JMC-2. 14 elements were outside a recovery range of 80%–120% for at least one of the analyzed reference materials.
Introduction
Levels of toxic heavy metals in the aquatic environment play a key role for the chemical and ecological quality status of habitats, which is reflected by the implementation of limit values of metals in a multitude of matrices covered by different directives (e.g. Water Framework Directive 2000/60/EC,1 Marine Strategy Framework Directive 2008/56/EG2 or the OSPAR3). Therefore, the permanent and extensive monitoring of metals is essential. In addition to the legacy metal pollutants (Ni, Cu, Zn, Cd, Hg, Pb), there is growing concern about different emerging metal contaminants, i.e. metal species such as Gd-based4 contrast agents or metal containing pharmaceuticals, as well as the so called technology-critical elements (TCE), which may become emerging pollutants in the near future due to their broad application, as well as missing technology for their recycling to support a circular economy.5–7 Due to the accumulation of metal contaminants over time, especially sediments may serve as time records/memories of aquatic systems' pollution by metals, posing a meaningful indicator to establish changes in rivers, estuaries and coastal areas and reflect the time-integrated pollution status.8,9Today, flame or electrothermal atomic absorption spectroscopy (FAAS, ETAAS), as well as plasma-based techniques like inductively coupled plasma-optical emission spectroscopy (ICP-OES) and especially inductively coupled plasma-mass spectrometry (ICP-MS) are most frequently used for multi-elemental analysis in environmental samples.10,11 For the analysis of sediment samples, the great majority of these methods require the dissolution of the analytes and herewith decomposition or leaching of the sample matrix.12 As of yet, various leaching protocols have been described in the literature e.g. based on NH3–HCl, acidified H2O2 or acetic acid, as proposed by the United Nations Environment Program for the extraction of heavy metal fraction from sediments originating from anthropogenic sources.13,14 However, leaching procedures will only determine the leachable, weak bound fraction of heavy metals, as the sample matrix (silicate) will not be decomposed. Therefore, the selected chemicals allow the mobilization of a specific element fraction. Consequently, elemental mass fractions determined by leaching procedures are not comparable to elemental mass fractions of total digestion protocols, as they serve different purposes.
Fusion approaches based on alkaline, acid and peroxide fusion using e.g. KNaCO3, KI, LiF, LiBO2/Li2B4O7, Li2CO3, Na2CO3, NaHCO3, NaNO3, NaHSO4, Na2O2, NaOH, NH4F·HF are frequently applied for dissolution of silicate-based matrices in geological applications.15–18 However, fusion techniques involve large quantities of the flux relative to the sample size. Therefore, impurities in the fusion salts pose a tremendous contamination source and may lead to elevated detection limits, as well as biased results. Moreover, high salt matrices pose a problem for atomic spectroscopy and mass spectrometric detection techniques (e.g. instability and high background readings, as well as interferences).
Acid digestions of silicate-based matrices involve the use of strong mineral acids like HNO3, HCl or HClO4 often in combination with HF. Today, closed-vessel/microwave-assisted digestion is considered superior to open-vessel/hot-plate based techniques for reasons like efficient, homogeneous and fast heat transfer, prevention of loss of volatile elements and compounds (chlorides and fluorides), as well as minimization of airborne contamination.19,20 Analyses of sediment reference materials via microwave-assisted digestion have shown that HF-based digestion protocols lead to significantly higher metal mass fractions (up to ∼20% for Cd and Cr) since it allows the mobilization of the element fraction bound to the silicate-matrix and other refractory oxides, that cannot be decomposed and dissolved by HNO3 and/or HCl.21 Therefore, digestions using aqua regia (e.g. DIN 38414 (1983) or ISO 11466, (1995)) can only provide pseudo total mass fractions.22 As the term pseudo total mass fractions implies a leaching with aqua regia may be sufficient to determine the total amount of selected elements. Indeed achievable recoveries highly depend on the analyte, as well as the matrix composition. Even though HF is considered highly hazardous and corrosive for all glass/quartz parts of analytical instruments, its usage is necessary to break down SiO2 compounds for different analytical applications.23 Besides difficulties in handling highly toxic HF, its use may also lead to considerable underestimation of a variety of other metals (e.g. Al, Ba, Ca, Mg and Se) due to formation of fluoride-based precipitates.24 Furthermore, the formation of insoluble fluorides may lead to the co-precipitation of other elements, as reported for the rare earth elements (REE).25 When using ICP-MS detection the entire sample introduction has to be changed to a PFA/Pt/Al/sapphire-based one (nebulizer, spray chamber, connectors, injector, torch) to avoid any damage due to the HF residues of the samples. Sample evaporation to remove the HF, as well as a reconstitution of the sample using HNO3 or aqua regia afterwards is a frequently applied approach, however this procedure is time consuming and loss of analytes has been observed. Often boric acid (H3BO3) is used to mask HF residues after total digestion, however this will again increase the matrix load of the digest, as well as the chance of sample contamination due to possible impurities of the used chemicals.
Alternative approaches to overcome difficulties in using/handling HF involve the use of in situ generation of HF by combination of fluoride-containing chemicals like NH4F or NH4F·HF with mineral acids.26 Formation of fluoride precipitation for these alternative digestion protocols is significantly lower than for conventional HF digests.27 Nevertheless, ammonium fluoride salts still require safe handling, as they are highly toxic. HF-free digestion protocols can also involve the use of H3PO4. Under high temperatures H3PO4 undergoes condensation reactions leading to the formation of condensed species (e.g. H4P2O7) capable of forming soluble SiP2O7.28 Although, chemicals of acute toxicity can be avoided with this approach, the use of H3PO4 leads to the formation of various polyatomic interferences in ICP-MS, thus hindering the detection of selected elements (e.g.63Cu (31P16O2), 64Zn (31P16O21H; 31P16O17O) or 87Sr (40Ar31P16O)).
The increasing number of metal contaminants of interest and the last decades' advances in mass spectrometry enabling time-efficient quantification of >50 metals in one run by ICP-MS/MS techniques demand for versatile and accurate sample preparation protocols. Thus, this work aims at the development and validation of an efficient protocol for complete microwave-assisted acid digestion of sediments using high-purity HBF4 as fluoride source, for subsequent analysis of >45 metals based on ICP-MS/MS. HBF4 is capable of in situ generation of HF, without being acutely toxic. An additional advantage of using HBF4 is that excess fluoride ions are directly complexed by H3BO3 and HBF3OH thus preventing precipitation of metal fluorides.25,29 This allows the direct analysis of the samples without any further measures, which helps to speed up sample preparation, as well as to avoid errors e.g. due to contamination or analyte loss. Despite the clear advantages of HBF4 over HF, as well as published applications of HBF4 based digestion protocols e.g. for the determination of rare earth elements25 and trace elements in peat30,31 or trace elements in bituminous sand mineral,32 its use is not widely recognized so far.
Experimental
Reagents and standards
All preparatory laboratory work was performed in a class 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000/1000 clean room. Type I reagent grade water (>18.2 MΩ cm) was obtained from an ultrapure water system consisting of an Elix 3 module (Merck Millipore, Darmstadt, Germany), a Milli-Q element module (Merck Millipore, Darmstadt, Germany) and a Q-POD element (Merck Millipore, Darmstadt, Germany). Analytical grade HNO3 (65% w/w, Fisher Scientific GmbH) and analytical grade HCl (30% w/w, Carl Roth GmbH + Co. KG, Karlsruhe, Germany) were further purified either by double sub-boiling in perfluoralkoxy-polymer (PFA)-subboling stills (DST-4000 & DST-1000, Savillex, Minnesota, USA) or by double sub-boiling using a cascade of two quartz stills (AHF Analysentechnik, Tübingen, Germany) operated under clean room conditions. HBF4 (38% w/w, Chem-Lab, Zedelgem, Belgium) and HF (Carl Roth GmbH, Karlsruhe, Germany) were used in ultra-pure quality for sample digestion without any further purification. For the initial development of the digestion method, HBF4 (48% w/w) with unknown elemental purity was used (Sigma Aldrich, Missouri, USA), as at this point of the work high purity HBF4 supplied by Chem-Lab was not yet available.
000/1000 clean room. Type I reagent grade water (>18.2 MΩ cm) was obtained from an ultrapure water system consisting of an Elix 3 module (Merck Millipore, Darmstadt, Germany), a Milli-Q element module (Merck Millipore, Darmstadt, Germany) and a Q-POD element (Merck Millipore, Darmstadt, Germany). Analytical grade HNO3 (65% w/w, Fisher Scientific GmbH) and analytical grade HCl (30% w/w, Carl Roth GmbH + Co. KG, Karlsruhe, Germany) were further purified either by double sub-boiling in perfluoralkoxy-polymer (PFA)-subboling stills (DST-4000 & DST-1000, Savillex, Minnesota, USA) or by double sub-boiling using a cascade of two quartz stills (AHF Analysentechnik, Tübingen, Germany) operated under clean room conditions. HBF4 (38% w/w, Chem-Lab, Zedelgem, Belgium) and HF (Carl Roth GmbH, Karlsruhe, Germany) were used in ultra-pure quality for sample digestion without any further purification. For the initial development of the digestion method, HBF4 (48% w/w) with unknown elemental purity was used (Sigma Aldrich, Missouri, USA), as at this point of the work high purity HBF4 supplied by Chem-Lab was not yet available.
External calibration standard solutions for quantification (all traceable to NIST standards) were prepared from custom-made multi-element standards of different composition (Inorganic Ventures, Christiansburg, USA).
The reference marine sediments NIST SRM 2702 (National Institute of Standards and Technology, Gaithersburg, USA), GBW 07313 (National Research Centre for Certified Reference Materials, Beijing, China) and JMS-2 (Geological Survey of Japan, Tokyo, Japan) as well as the reference stream sediment GBW 07311 (National Research Centre for Certified Reference Materials) were used for method development and validation.
Sediment digestion
50 mg aliquots of the sediment samples were digested either with a MARS Xpress or a MARS 6 (CEM Corp., Kamp Lintfort, Germany) microwave system. Digestion took place at 180 °C for 300 min using 35 mL pre-cleaned TFM digestion vessels. In total, six different acid mixtures were applied for sediment digestion: 2 mL HNO3/6 mL HCl; 5 mL HNO3/1 mL HF; 5 mL HNO3/2 mL HCl/1 mL HF; 5.5 mL HNO3/2 mL HCl/0.5 mL HBF4; 5 mL HNO3/2 mL HCl/1 mL HBF4 and 4 mL HNO3/2 mL HCl/2 mL HBF4. The corresponding acid volumes of each acid composition (total volume of 8 mL) were chosen based on the manufacturer recommendations. It should be noted that higher acid volumes will lead to higher pressures during digestion, which may consequently lead to a pressure release during digestion, including a possible loss of analyte. On the other hand, we wanted to make sure to use an acid volume as high as possible in order to prevent a shortage of acid. After digestion, the clear sample solutions were quantitatively transferred to 50 mL pre-cleaned DigiTUBEs (SCP Science, Quebec, Canada) and diluted to a final volume of 50 mL with type I reagent grade water. After each digestion, all vessels were cleaned by running three blank digests using a HNO3/HCl mixture. Digestions containing HF were evaporated to dryness using an evaporation unit (XpressVap, CEM Corp., Kamp Lintfort, Germany). HF-containing fumes were neutralized using a cascade of gas washing bottles containing Milli-Q water and KOH solution, respectively. After evaporation, the samples were re-dissolved in 2 mL concentrated HCl and 4 mL type I reagent grade water using a MARS Xpress. A detailed description of the used microwave parameters is shown in Table 1.| Microwave program for sediment dissolution (temperature controlled) | |||
|---|---|---|---|
| Power [W] | Temperature [°C] | Ramp. [min] | Hold [min] |
| 1600 | 180 | 60 | 240 |
| Microwave program for evaporation of HF containing digests (power controlled) | ||
|---|---|---|
| Power [W] | Hold [min] | Hold [min] |
| 800 | 90 | 90 |
| Microwave program for re-dissolution of HF containing digests (temperature controlled) | |||
|---|---|---|---|
| Power [W] | Temperature [°C] | Ramp. [min] | Hold [min] |
| 1600 | 80 | 20 | 60 |
Instrumentation and measurement procedures
Determination of elemental concentrations in the sediment digests was performed using an ICP-MS/MS (Agilent 8800, Agilent Technologies, Tokyo, Japan) coupled to an ESI SC-4 DX FAST autosampler (Elemental Scientific, Omaha, Nebraska, USA) equipped with a discrete sampling system with a loop volume of 1.5 mL. A list of measured isotopes and their detection modes, as well as a detailed description of all ICP-MS/MS operating parameters and used cell gas modes can be found in the ESI Table A1 and Table A2,† respectively. In total four different cell gas modes were used: no gas, He, H2 and O2, of which O2 was applied as reaction gas in MS/MS mode. Selection of the analyzed isotope, as well as cell gas mode was based on the achieved sensitivity and occurrence of spectral interferences (isobaric, as well as polyatomic interferences). In depth, extensive reviews about the working principles of ICP-MS/MS and potential isobaric interferences can be found in the literature.33,34 Based on the presented set-up the analysis of one sample took approx. 240 seconds. The instrument was optimized on a daily basis using a tune solution containing Li, Co, Y, Ce, and Tl (10 μg L−1). Quantification was performed based on external calibration, covering a concentration range from 0.1 μg L−1 to 100 μg L−1 for all analytes. Solutions were prepared on a daily basis from custom made multi-element standards (Inorganic Ventures, Christiansburg, USA). Wash blanks of 2% (w/w) HNO3 were measured after each sample triplicate to monitor potential carry-over effects.Data processing and calculations
Multi-element data were processed using MassHunter versions 4.2 to 4.4 (Agilent Technologies, Tokyo, Japan) and a custom-written Excel© spreadsheet. Limits of detection (LOD) (3 × SD) and limits of quantification (LOQ) (10 × SD) of the method were calculated in accordance with MacDougall et al. from procedural blanks (n = 3).35 Combined uncertainties with a coverage factor of 2 (U, k = 2) were calculated taking into account reproducibility of multiple digests and measurement precision of the samples.The significant number of digits of mass fractions are given according to GUM and EURACHEM guidelines, whereby the uncertainty determines the significant number of digits to be presented with the value.36,37
Zeta sores (ζ) that are usually applied in interlaboratory studies were adapted, in order to provide an easy to grasp overview over the performance of the digestion protocol for each analyte. The zeta sores were calculated according to following equation, with xlab measured mass fraction, Xref certified mass fraction, Ulab expanded uncertainty of the measured mass fraction and Uref expanded uncertainty of certified mass fraction. Similar to z scores zeta scores outside ±2 are commonly regarded as questionable. Indeed, if expanded uncertainties are used instead of standard uncertainties zeta scores will only be about one half, leading to the fact that values outside ±1 should be regarded questionable.38
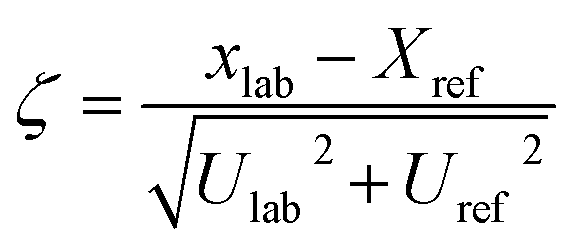
Results and discussion
Based on initial experiments, the efficiency of the digest was evaluated in terms of sample weight, temperature and duration of the digestion. For this purpose, samples of 50 mg and 100 mg of NIST SRM 2702 were digested for three hours at a temperature of 180 °C. The resulting data indicates that with the time setting and the amount of acid used, a sample weight of 50 ± 5 mg results in better overall recovery. Therefore, all further experiments were conducted with a sample weight of 50 mg. Experiments showed that temperatures higher than 180 °C may be achieved with the used microwave set-up, but at the cost of evaporation of the digest, due to a possible pressure release during digestion. Since the digestion temperature was limited to 180 °C, it was decided to extend the duration of the digestion to 300 minutes, in order to ensure a quantitative digestion of the matrix.Recoveries of NIST SRM 2702 for digestion protocols based on HF
In order to develop a quantitative digestion protocol for a large variety of analytes, the marine sediment reference material NIST SRM 2702 was used for method development. This reference material is certified for a large variety of analytes and can be purchased from the National Institute of Standards & Technology (NIST). Fig. 1 shows recoveries for certified elements using aqua regia, a mixture of HNO3 and HF, as well as a mixture of HNO3, HCl and HF for digestion.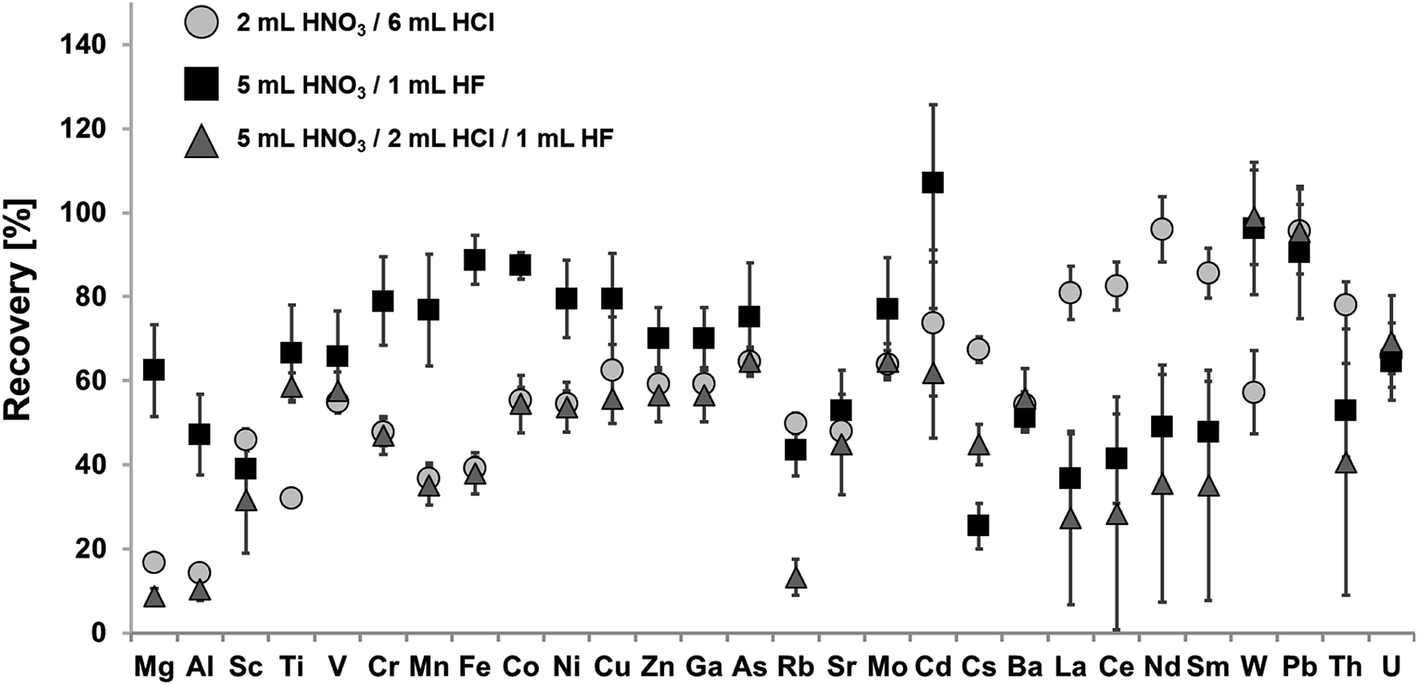 | ||
| Fig. 1 Recoveries for the digestion of 50 mg of NIST SRM 2702 using different acid mixtures (2 mL HNO3/6 mL HCl (circle); 5 mL HNO3/1 mL HF (square); 5 mL HNO3/2 mL HCl/1 mL HF (triangle)) digested at 180 °C for 300 min. Error bars correspond to expanded uncertainties U (k = 2), n = 3; if not visible they are in the range of spot size. | ||
The aim of this first step of method development was the application and comparison of achievable recoveries of different commonly used acid compositions. This includes strongly oxidizing aqua regia, which may be used to determine “pseudo total” mass fractions for some analytes, e.g. according to ISO 11466:1995 (soil quality – extraction of trace elements soluble in aqua regia). Other commonly used acid mixtures for the digestion of geological samples comprise of HNO3 and HF in varying mixing ratios. HNO3 is a strong oxidizing agent combined with the capability of HF to dissolve silicate and other refractory oxides. The last acid composition applies HCl in combination with HNO3 and HF. Hereby, the complexing ability of Cl− may help to improve recoveries for some elements. Furthermore, HCl is capable of digesting many metal oxides.
Achieved recoveries for the aqua regia digest (2 mL HNO3 and 6 mL HCl) ranged from 14.3% ± 1.4% for Al to 96% ± 8% for Nd, for digests using a mixture of HNO3 and HF (5 mL/1 mL) from 25% ± 5% for Cs to 107% ± 19% for Cd and for digests using a mixture of HNO3, HCl and HF (5 mL/2 mL/1 mL) from 8.8% ± 1.6% for Mg to 99% ± 11% for W.
For some analytes, a leaching with aqua regia is already sufficient for their quantitative extraction. This is true for selected heavy metals e.g. Pb (recovery: 96% ± 6%) or the REE e.g. Nd (recovery: 96% ± 8%). Therefore, treatment with aqua regia is a commonly used method for the determination of selected heavy metals in sediments, e.g. when looking for the maximum bioavailable fraction. Nevertheless, leaching efficiency highly depends on the matrix composition of the digested sediments, as aqua regia does not dissolve silicates and other refractory oxides. Results might therefore be prone to error. As a result, a pure leaching is not sufficient for a multi-element analysis, when aiming for the fingerprinting of the total elemental composition of the analyzed sediment sample.
Significantly higher recoveries for transition metals (Co, Ni, Cu) can be achieved by using an acid mixture consisting of HNO3 and HF. Besides, the dissolution of the silicate matrix, HF will also lead to the dissolution of refractory oxides like Cr2O3 and Fe2O3, thus achieving significantly higher recoveries for these metals in comparison to a pure leaching with aqua regia. On the other hand, the formation of insoluble fluoride precipitates, mainly consisting of Mg, Al, Ca and Na fluorides, during HF digestions has been described in the literature.25 Therefore, co-precipitation of certain elements like Rb, Sr, Cs, Ba, REE or Th may occur. This hinders the accurate determination of these elements, as displayed in Fig. 1. One way to suppress this effect may be the addition of Mg. Takei et al. attributed a reduced fluoride formation to the ratio of Mg and Ca to Al.39 However, this procedure has the disadvantage that Mg cannot be an analyte and it additionally introduces a risk of contamination due to impurities of the used Mg salt.
Similar observations can be made for an acid mixture containing HNO3, HCl and HF. Again, recoveries especially for the REE are not quantitative. Furthermore, overall recoveries for almost all elements are significantly lower than for the acid composition containing only HNO3 and HF. During this work, digests containing HF were evaporated and re-dissolved prior to analysis via ICP-MS, which is necessary in order to remove unreacted HF from the sample solution. Indeed, this step may also lead to the loss of volatile analytes such as Hg.
Taking into account the results of the applied acid mixtures, it was decided to proceed the method development with an acid mixture of 5 mL HNO3 and 2 mL HCl for the following replacement of HF with HBF4. Even though, the acid mixture of HNO3 and HF (5 mL/1 mL) achieved higher recoveries for some analytes than the acid mixture of HNO3, HCl and HF (5 mL/2 mL/1 mL). The addition of HCl is assumed unavoidable for a digestion protocol aiming at the accurate determination of a wide variety of analytes, due to the complexing abilities of Cl− as most chloride complexes are very stable in aqueous solution.40
Digestion protocol based on HBF4
Fig. 2 shows recoveries for certified elements in NIST SRM 2702 using a mixture of HNO3, HCl and varying amounts of HBF4 for digestion. Achieved recoveries for the digest using 0.5 mL of HBF4 ranged from 58% ± 7% for Sc to 158% ± 20% for As, for digests using 1 mL of HBF4 from 56% ± 11% for Sc to 237% ± 49% for As and for digests using 2 mL HBF4 from 45% ± 1% for Mg to 426% ± 40% for As.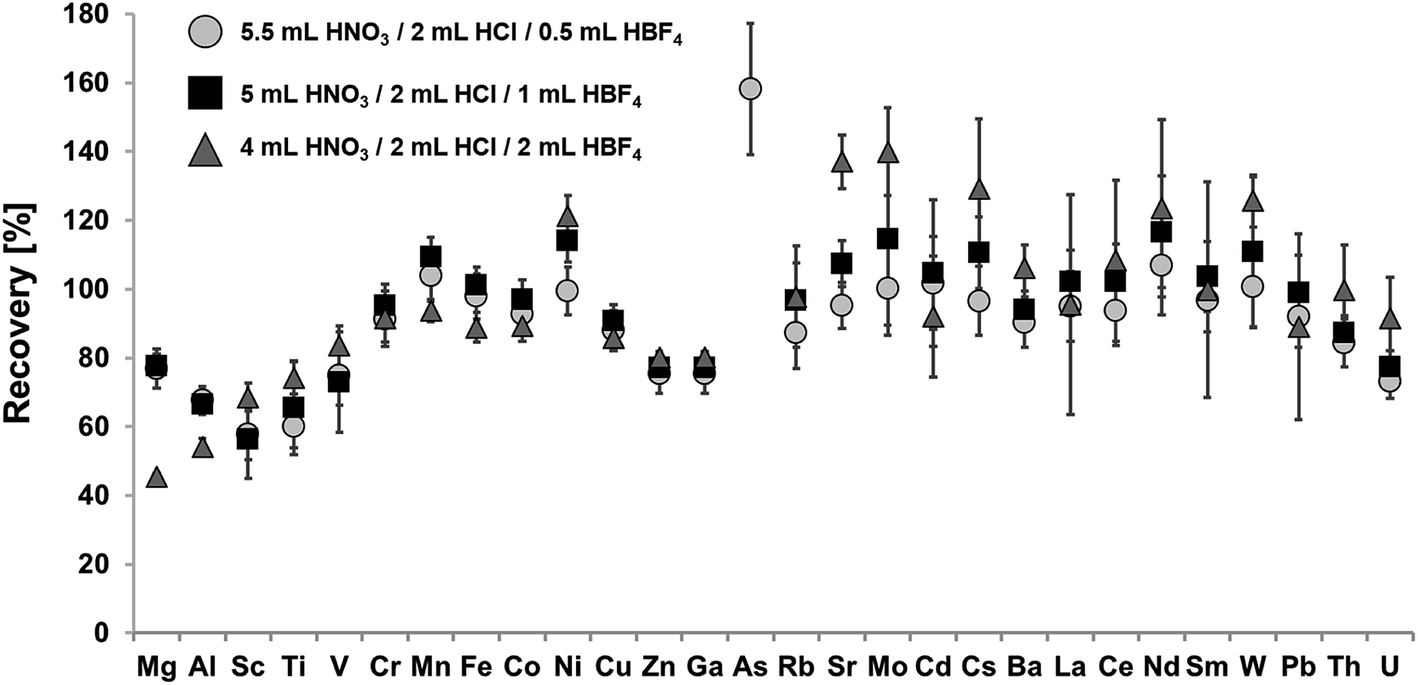 | ||
| Fig. 2 Recoveries for 50 mg of NIST SRM 2702 sediment for different HBF4 containing acid mixtures (5.5 mL HNO3/2 mL HCl/0.5 mL HBF4 (circle); 5 mL HNO3/2 mL HCl/1 mL HBF4 (square); 4 mL HNO3/2 mL HCl/2 mL HBF4 (triangle)) digested at 180 °C for 300 min. Values of As for the acid mixtures 5 mL HNO3/2 mL HCl/1 mL HBF4 (square) and 4 mL HNO3/2 mL HCl/2 mL HBF4 (triangle) are not shown (recoveries: 237% ± 49% and 426% ± 40%). Error bars correspond to expanded uncertainties U (k = 2), n = 3; if not visible they are in the range of spot size. | ||
All used acid mixtures containing HBF4 achieved significantly higher recoveries and precision than the afore described acid mixtures. Recoveries were >80% (within uncertainty) for all analyzed elements with the exception of Mg, Al, Sc, Ti, V, Zn, Ga and U. The formation of insoluble fluoride precipitates is significantly lower than for HF-containing digestion protocols, leading to recoveries for Mg of up to 78% ± 4%. Furthermore, co-precipitation of e.g. REE can be avoided, leading to quantitative REE recoveries. Nevertheless, co-precipitation may become significant for high amounts of HBF4, as shown for the acid mixture containing 2 mL HBF4, leading to low recoveries for Mg and Al (45% ± 1%, 54% ± 3%). The use of HBF4 also enables the digestion refractory oxides like Cr2O3 and Fe2O3, resulting in quantitative recoveries for both elements for all acid mixtures (>91% ± 7% Cr, 89% ± 4% Fe). As shown in Table 2, low purity HBF4 may contain significant amounts of e.g. Mn, As, Sr and Mo, which may significantly bias the recoveries of these elements. This is evident in case of As for which recoveries ranged between 158% ± 20% to 426% ± 40%. Therefore, blank contributions of all potential analytes should be monitored on a regular basis.
| Element | LOQ [μg L−1] unknown purity HBF4 | Blank contribution [%] | LOQ [μg L−1] ultra pure HBF4 | Blank contribution [%] | Element | LOQ [μg L−1] unknown purity HBF4 | Blank contribution [%] | LOQ [μg L−1] ultra pure HBF4 | Blank contribution [%] |
|---|---|---|---|---|---|---|---|---|---|
| Mg | 85 | 0.9 | 1.9 | 0.019 | Rb | 0.15 | 0.12 | 0.02 | 0.016 |
| Al | 32 | 0.04 | 6.7 | 0.008 | Sr | 22 | 18 | 0.02 | 0.016 |
| Sc | 0.4 | 1.6 | 0.05 | 0.17 | Mo | 3 | 31 | 0.02 | 0.15 |
| Ti | 250 | 2.8 | 0.2 | 0.0024 | Cd | 0.02 | 2.1 | 0.011 | 1.3 |
| V | 0.5 | 0.13 | 0.03 | 0.007 | Cs | 0.9 | 12 | 0.02 | 0.3 |
| Cr | 11 | 3 | 0.3 | 0.10 | Ba | 2.1 | 0.5 | 0.014 | 0.003 |
| Mn | 156 | 9 | 0.07 | 0.004 | La | 0.04 | 0.06 | 0.004 | 0.006 |
| Fe | 94 | 0.13 | 0.2 | 0.0003 | Ce | 0.09 | 0.08 | 0.005 | 0.004 |
| Co | 0.14 | 0.5 | 0.017 | 0.06 | Nd | 0.09 | 0.16 | 0.006 | 0.011 |
| Ni | 12 | 15 | 0.04 | 0.06 | Sm | 0.01 | 0.14 | 0.005 | 0.05 |
| Cu | 2.4 | 2.0 | 0.11 | 0.10 | W | 0.6 | 10 | 0.03 | 0.4 |
| Zn | 2.7 | 0.6 | >0.48 | >0.10 | Pb | 0.3 | 0.19 | 1.2 | 0.9 |
| Ga | 0.7 | 2.9 | >0.005 | >0.021 | Th | 0.2 | 1.2 | 0.08 | 0.4 |
| As | 84 | 185 | 0.7 | 1.5 | U | 0.5 | 5 | 0.002 | 0.018 |
In conclusion, the mixture of 5 mL HNO3, 2 mL HCl and 1 mL HBF4 has been shown to give the most consistent results for the broadest number of elements. Hence, this acid mixture was used for all following considerations.
Achievable LOQ for HBF4 containing digests
As can be seen in Fig. 2, recoveries for some analytes significantly increase above 100% with increasing HBF4 amounts e.g. As with 426% ± 42%, Sr with 137% ± 8% and Mo with 140% ± 13% for the digestion mixture using 2 mL of HBF4. This increase can be assigned to impurities of the used HBF4. In fact, to the authors knowledge, there is currently only one supplier of high purity HBF4. Unfortunately, high purity HBF4 was not available for the initial experiments conducted in the previous two chapters. However, this allowed for the comparison of achievable LOQs for two different HBF4 suppliers, as shown in Table 2. Furthermore, Table 2 shows the corresponding blank contribution in % based on the analysis of 50 mg SRM 2702. This will facilitate an easy comparison of possible blank contributions influencing the elemental recovery for each element.As enumerated in Table 2, main impurities of the used HBF4 are Mg, Al, Ti, Mn, Fe, As and Sr. Since Mg, Al, Ti and Fe are main matrix components (% range), possible contaminations are only of minor concern. E.g. the blank contribution of Ti for HBF4 with low purity is 2.8%, which is in the range of the measurement uncertainty, but still significantly lower than the combined uncertainty. Nevertheless, significantly higher recoveries (>100%) were observed for elements like Ni, As and Sr or Mo which are clearly associated with high blank levels of these elements. The blank contribution (Ni: 15%, As: 185%, Sr: 18%, Mo: 31%) of these elements significantly influence the achieved recoveries, thus leading to wrong results. High purity HBF4 blank levels of these elements are significantly lower, thus minimizing possible contamination. The highest blank contribution of 1.5% for use of the high purity HBF4 was found for As. With the exception of Cd, Cs, W, Pb and Th blank contributions of all other analyzed elements are lower than 0.2%. Even if the use of low purity HBF4 is sufficient for correct quantification of many elements, a universal multi-elemental digestion protocol clearly benefits from high purity HBF4. If high purity HBF4 is not available, careful blank monitoring of all analytes is advisable.
Reference materials digested with optimized digestion protocol
The developed digestion protocol using 5 mL HNO3, 2 mL HCl and 1 mL high purity HBF4 was applied for the analysis of different other sediment reference materials (GBW 07313, GBW 07311 and JMS-2). Results for all analyzed elements can be found in Table 3. All three reference materials were quantitatively digested by the optimized digestion protocol, resulting in clear particle-free digests.| GBW 07313 (n = 35) | GBW 07311 (n = 17) | JMS-2 (n = 18) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Element | Measured [mg kg−1] | Certified range [mg kg−1] | Recovery [%] | Measured [mg kg−1] | Certified range [mg kg−1] | Recovery [%] | Measured [mg kg−1] | Certified range [mg kg−1] | Recovery [%] |
| Be | 1.8 ± 0.6 | 25 ± 8 | 26 ± 3 | 95 | 1.6 ± 0.3 | 1.8 ± 0.1 | 90 | ||
| Mg | 19000 ± 5000 |
20400 ± 300 |
93 | 3300 ± 800 | 3700 ± 400 | 89 | 16000 ± 3000 |
19540 ± 120 |
80 |
| Al | 69000 ± 13000 |
72800 ± 500 |
95 | 40000 ± 26000 |
54900 ± 500 |
72 | 53000 ± 26000 |
75000 ± 500 |
70 |
| P | 1500 ± 500 | 2000 ± 100 | 75 | 219 ± 26 | 255 ± 27 | 86 | 5800 ± 500 | 5500 ± 90 | 105 |
| K | 24000 ± 5000 |
24500 ± 400 |
98 | 28000 ± 5000 |
27200 ± 600 |
101 | 18800 ± 2700 |
22410 ± 170 |
84 |
| Ca | 3400 ± 700 | 3360 ± 210 | 100 | 32000 ± 4000 |
33500 ± 400 |
97 | |||
| Sc | 27 ± 5 | 25.6 ± 2.9 | 105 | 4 ± 4 | 7.4 ± 0.4 | 50 | 18 ± 13 | ||
| Ti | 4000 ± 900 | 4000 ± 100 | 100 | 2000 ± 700 | 2100 ± 100 | 97 | 8100 ± 700 | 8391 ± 120 | 97 |
| V | 110 ± 18 | 112 ± 5 | 98 | 49 ± 6 | 47 ± 3 | 104 | 187 ± 14 | 183 ± 3 | 102 |
| Cr | 61 ± 10 | 58.4 ± 1.3 | 104 | 44 ± 8 | 40 ± 3 | 109 | 77 ± 8 | 78 ± 1 | 98 |
| Mn | 3300 ± 700 | 3300 ± 100 | 100 | 2700 ± 400 | 2490 ± 80 | 110 | 16000 ± 1600 |
17500 ± 160 |
91 |
| Fe | 45000 ± 8000 |
46000 ± 500 |
98 | 32000 ± 5000 |
30700 ± 500 |
106 | 68000 ± 700 |
76700 ± 600 |
89 |
| Co | 78 ± 14 | 76.7 ± 1.2 | 102 | 9.3 ± 1.1 | 8.5 ± 0.8 | 110 | 224 ± 13 | 226 ± 2 | 99 |
| Ni | 150 ± 28 | 150 ± 4 | 100 | 16.9 ± 2.1 | 14.3 ± 1.0 | 118 | 301 ± 21 | 311 ± 3 | 97 |
| Cu | 390 ± 70 | 424 ± 8 | 92 | 80 ± 10 | 79 ± 3 | 102 | 368 ± 22 | 447 ± 2 | 82 |
| Zn | 150 ± 25 | 160 ± 3 | 94 | 333 ± 40 | 373 ± 14 | 89 | 123 ± 7 | 166 ± 2 | 74 |
| Ga | 28 ± 5 | 23.7 ± 1.7 | 118 | 20.5 ± 2.2 | 18.5 ± 0.9 | 111 | 25 ± 4 | ||
| As | 10.0 ± 1.4 | 5.8 ± 0.8 | 172 | 217 ± 29 | 188 ± 13 | 116 | 39 ± 4 | 35 ± 1 | 112 |
| Rb | 100 ± 17 | 97.3 ± 2.6 | 103 | 490 ± 110 | 408 ± 11 | 124 | 65 ± 22 | 65 ± 1 | 99 |
| Sr | 260 ± 60 | 267 ± 15 | 97 | 35 ± 4 | 29 ± 4 | 119 | 491 ± 30 | 454 ± 4 | 108 |
| Zr | 150 ± 23 | 177 ± 10 | 85 | 70 ± 17 | 153 ± 13 | 46 | 210 ± 18 | 220 ± 3 | 95 |
| Mo | 7.3 ± 1.1 | 7.2 ± 0.5 | 101 | 7.2 ± 0.8 | 5.9 ± 0.6 | 121 | 23 ± 2 | ||
| Ag | 0.11 ± 0.03 | 3.2 ± 0.3 | 3.2 ± 0.4 | 99 | 0.26 ± 0.03 | ||||
| Cd | 0.38 ± 0.09 | 2.2 ± 0.5 | 2.3 ± 0.2 | 94 | 0.44 ± 0.07 | ||||
| Sb | 2.5 ± 0.5 | 1.9 ± 0.4 | 135 | 23 ± 3 | 14.9 ± 1.2 | 154 | 5.6 ± 0.5 | 4.5 ± 0.2 | 124 |
| Te | 0.32 ± 0.10 | 0.7 ± 0.5 | 0.40 ± 0.10 | 181 | 1.5 ± 0.7 | 1.38 ± 9 | 109 | ||
| Cs | 9.1 ± 1.4 | 9.4 ± 0.7 | 97 | 21 ± 3 | 17 ± 1 | 120 | 3.5 ± 0.3 | 3.0 ± 0.2 | 116 |
| Ba | 4500 ± 1000 | 4400 ± 200 | 102 | 260 ± 340 | 260 ± 17 | 102 | 1881 ± 124 | 1856 ± 16 | 101 |
| La | 71 ± 10 | 67.8 ± 2.9 | 105 | 23 ± 10 | 30 ± 2 | 79 | 107 ± 52 | ||
| Ce | 96 ± 15 | 92 ± 8 | 104 | 50 ± 16 | 58 ± 4 | 86 | 133 ± 48 | ||
| Pr | 22 ± 3 | 20.1 ± 1.9 | 109 | 6.3 ± 2.2 | 7.4 ± 0.5 | 85 | 31 ± 9 | ||
| Nd | 93 ± 13 | 92 ± 4 | 101 | 24 ± 8 | 27 ± 2 | 89 | 135 ± 38 | ||
| Sm | 22 ± 3 | 21.5 ± 1.3 | 102 | 7.2 ± 2.5 | 6.2 ± 0.3 | 116 | 38 ± 10 | ||
| Eu | 5.8 ± 0.9 | 5.3 ± 0.3 | 109 | 0.7 ± 0.3 | 0.60 ± 0.06 | 124 | 10.1 ± 2.4 | ||
| Gd | 23 ± 3 | 22.0 ± 1.2 | 105 | 7.2 ± 2.8 | 5.9 ± 0.4 | 121 | 45 ± 12 | ||
| Tb | 3.6 ± 0.5 | 3.4 ± 0.3 | 106 | 1.2 ± 0.5 | 1.13 ± 0.09 | 108 | 7.2 ± 1.6 | ||
| Dy | 21 ± 3 | 19.9 ± 1.8 | 106 | 7 ± 3 | 7.2 ± 0.6 | 101 | 43 ± 9 | ||
| Ho | 4.1 ± 0.6 | 4.3 ± 0.2 | 95 | 1.4 ± 0.6 | 1.4 ± 0.2 | 98 | 8.6 ± 1.6 | ||
| Er | 12.0 ± 1.6 | 11.0 ± 0.7 | 109 | 4.1 ± 1.8 | 4.6 ± 0.5 | 90 | 25 ± 4 | ||
| Tm | 1.60 ± 0.22 | 1.54 ± 0.14 | 104 | 0.6 ± 0.3 | 0.74 ± 0.09 | 86 | 3.3 ± 0.5 | ||
| Yb | 10.00 ± 1.4 | 9.8 ± 1.1 | 102 | 4.3 ± 1.9 | 5.1 ± 0.6 | 85 | 21 ± 4 | ||
| Lu | 1.60 ± 0.22 | 1.46 ± 0.19 | 110 | 0.6 ± 0.3 | 0.78 ± 0.06 | 82 | 3.3 ± 0.5 | ||
| W | 5.7 ± 0.8 | 5.5 ± 0.6 | 104 | 133 ± 16 | 126 ± 9 | 106 | 6.2 ± 0.5 | ||
| Tl | 0.97 ± 0.20 | 2.9 ± 0.4 | 2.9 ± 0.4 | 99 | 2.67 ± 0.18 | ||||
| Pb | 29 ± 8 | 29.3 ± 1.1 | 99 | 690 ± 150 | 636 ± 22 | 111 | 84 ± 12 | 88 ± 2 | 96 |
| Bi | 0.92 ± 0.19 | 63 ± 9 | 50 ± 4 | 126 | 1.45 ± 0.09 | ||||
| Th | 14 ± 4 | 13.9 ± 1.1 | 101 | 22 ± 10 | 23.3 ± 1.2 | 95 | 11 ± 10 | ||
| U | 1.7 ± 0.3 | 2.0 ± 0.5 | 86 | 10.2 ± 1.4 | 9.1 ± 0.9 | 112 | 2.92 ± 0.21 | ||
In order to give a first overview over the overall performance of the developed digestion protocol Table 4 presents zeta scores for all certified elements of the three analyzed reference materials. Within this context a value outside of ±1 is usually regarded as questionable, as in this case zeta scores were calculated based on combined uncertainties.38 It should be emphasized that proficiency testing tools like the zeta score are usually used within a different context e.g. for long term interlaboratory studies, as zeta scores may easily demonstrate the performance of a laboratory/method. However, a low zeta score does not necessarily indicate good quality of analytical results, by stating an unrealistically high uncertainty zeta scores may be artificially decreased. Therefore, zeta scores presented here are meant to give a first comprehensive overview in order to easily identify the performance for each analyte of the developed digestion protocol. For elemental mass fractions certified in all three analyzed reference materials only zeta scores for Sb were outside ±1 for all three materials. For the elements As and Cs, two of three reference materials were outside the range of ±1. Considering the analysis of 48 elements in total, the presented zeta scores already underpin the multi-elemental capabilities of the developed digestion protocol.
| Zeta-score ζ | |||
|---|---|---|---|
| Element | GBW 07313 (n = 35) | GBW 07311 (n = 17) | JMS-2 (n = 18) |
| Be | −0.15 | −0.49 | |
| Mg | −0.30 | −0.45 | −1.18 |
| Al | −0.29 | −0.57 | −0.85 |
| P | −0.96 | −0.97 | 0.59 |
| K | −0.11 | 0.16 | −1.33 |
| Ca | 0.05 | −0.37 | |
| Sc | 0.24 | −0.84 | |
| Ti | 0.00 | −0.14 | −0.41 |
| V | −0.11 | 0.27 | 0.25 |
| Cr | 0.26 | 0.45 | −0.15 |
| Mn | 0.00 | 0.56 | −0.93 |
| Fe | −0.12 | 0.34 | −9.44 |
| Co | 0.09 | 0.60 | −0.19 |
| Ni | 0.00 | 1.15 | −0.46 |
| Cu | −0.52 | 0.14 | −3.52 |
| Zn | −0.40 | −0.95 | −5.92 |
| Ga | 0.76 | 0.84 | |
| As | 2.60 | 0.93 | 1.09 |
| Rb | 0.16 | 0.74 | −0.02 |
| Sr | −0.12 | 0.96 | 1.20 |
| Zr | −1.08 | −3.92 | −0.58 |
| Mo | 0.08 | 1.24 | |
| Ag | −0.06 | ||
| Cd | −0.27 | ||
| Sb | 1.07 | 2.87 | 2.14 |
| Te | 0.70 | 0.18 | |
| Cs | −0.19 | 1.35 | 1.23 |
| Ba | 0.10 | 0.00 | 0.20 |
| La | 0.31 | −0.67 | |
| Ce | 0.24 | −0.45 | |
| Pr | 0.52 | −0.50 | |
| Nd | 0.09 | −0.36 | |
| Sm | 0.16 | 0.39 | |
| Eu | 0.55 | 0.55 | |
| Gd | 0.28 | 0.45 | |
| Tb | 0.34 | 0.19 | |
| Dy | 0.32 | 0.02 | |
| Ho | −0.34 | −0.05 | |
| Er | 0.57 | −0.26 | |
| Tm | 0.23 | −0.36 | |
| Yb | 0.11 | −0.40 | |
| Lu | 0.48 | −0.50 | |
| W | 0.20 | 0.40 | |
| Tl | −0.04 | ||
| Pb | −0.04 | 0.36 | −0.30 |
| Bi | 1.35 | ||
| Th | 0.03 | −0.12 | |
| U | −0.48 | 0.67 | −0.49 |
This is also reflected in almost quantitative recoveries for the main components Mg, Al, P, K, Ca, and Fe (<70%), even though relative combined uncertainties of the results are significantly higher than for trace components. Measured mass fractions of “classical” heavy metals such as Co, Ni, Cu, Zn, Cd or Pb all overlapped within the stated uncertainties with the certified values (except Cu and Zn for JMS-2). The use of HBF4 as fluorine source also achieves the digestion of refractory oxides like Cr2O3 and ZrO2, which are challenging to dissolve. Measured mass fractions of Cr and Zr overlapped within the stated uncertainties with the certified values (except Zr for GBW 07311).
The quantitative recovery of elements like Rb, Sr, Ba, Cs, which are known to suffer from non-quantitative recoveries due to fluoride co-precipitation, indicates no formation of insoluble fluorides during the presented digestion protocol, which can be explained by the equilibrium between HBF4, free HF and free H3BO3 during digestion. Moreover, this also results in quantitative recovery of all REE.
Conclusion
With the continuously improving multi-element capabilities of modern ICP-MS(/MS) instruments the quasi-simultaneous analysis of most elements of the periodic table became possible. Besides the analysis of classical heavy metals such as Cu, Zn, Cd and Pb the same sample can now be analyzed for a large set of elements without significantly increasing analysis time, which opens challenging new research questions in various scientific fields, e.g. the analysis of TCE in environmental matrices or multi-element trace and fingerprint analysis. However, this requires validated sample preparation protocols specifically designed for such kind of multi-element analysis.Sample preparation, including sample dissolution can still be regarded as the bottleneck during elemental analysis. Up to now, the majority of digestion protocols is based on the use of HF, in order to dissolve the silicate matrix and refractory oxides. Nevertheless, loss of specific analytes due to fluoride co-precipitation have been reported, making it difficult to establish multi-elemental extraction procedures for specific analytes. Furthermore, the acute toxicity of HF requires careful sample handling or its use is restricted in many labs which requires additional sample preparation steps like evaporation and re-dissolution. The presented digestion protocol uses the less dangerous and non-acute toxicity source of fluoride HBF4, thus risks of handling HF-containing solutions is greatly decreased. During digestion, HF is generated in situ and excess fluoride ions are directly complexed by in situ generated H3BO3. Therefore, digests can be directly measured e.g. via ICP-MS without the need of further sample preparation steps or HF resistant sample introduction systems. Based on the same reaction the formation of fluoride precipitates is also minimized, as free HF during digestion either reacts with the sample or is being neutralized by H3BO3 to form HBF4 again. Thus, sample digestion based on HBF4 is suitable for the quantification of a large variety of elements with a minimum of sample preparation steps.
In comparison to other multi-elemental digestion protocols based on the use of e.g. a combination of HNO3, HCl and HF, LOQs of the presented HBF4 based digestion protocol are comparable (e.g. Ni: 0.016 μg L−1vs. 0.04 μg L−1 for this study) or even lower (e.g. Fe: 1.70 μg L−1vs. 0.2 μg L−1 for this study).41 Similar studies dedicated to the multi-elemental analysis of sediment reference materials achieved comparable mass fractions for a large variety of analytes. E.g. Fiket et al. analyzed 46 elements in sediment and soil reference materials including the reference material GBW 07311 analyzed in this study. For 31 analyzed elements of the reference material GBW 07311 recoveries ranged from 90% (Tl) to 104% (Ag), based on the use of 4 mL HNO3, 1 mL HCl and 1 mL HF.42 Roje et al. analyzed the mass fractions of 26 elements of the reference material GBW 07311 based on different acid composition (HNO3, aqua regia and a mixture of HNO3, HCl and HF) and reported recoveries ranging from 9% (Sn) to >100% (Ag, Be, Bi, Li, Mn, Pb, Ti).41
As a future application, the new digestion protocol may also be used for the dissolution of geological samples for isotope ratio analysis e.g. Sr, Nd or Pb in isotope geochemistry. As a proof of principle, the presented digestion protocol has already been applied within this context.43 Furthermore, large-scale environmental studies analyzing e.g. sediment samples will benefit from easier sample preparation and improvements in analysis and sample preparation time. Therefore, the presented study proposes use of HBF4 as alternative to HF for the digestion of sediments during environmental analysis. Specifically, a mixture of 5 mL HNO3, 2 mL HCl and 1 mL HBF4, which allows the routine microwave-assisted digestion of a 50 mg sediment sample, which is sufficient for multi-element analysis as demonstrated for a variety of reference materials.
Future studies should also further evaluate the suitability of the substitution of HF by HBF4 for the digestion of other geological matrices like different types of rock samples, soil samples or dusts.
Conflicts of interest
The authors have no competing interests to declare.Acknowledgements
The authors thank Nikolai Huwa, Maike Schmidt, Andrea Lapschies, Bettina Rust and Nathalie Voigt for their support in the laboratory. Anna Reese was funded by the BSH through the project OffChEm (BSH contract code: 10036781, HZG contract code: 17/2017).References
- Europaen Commission, Directive 2000/60/EC of the European Parliament and of the Council of 23 October 2000 establishing a framework for Community action in the field of water policy, Official Journal of the European Communities, 2000 Search PubMed.
- Europaen Commission, Directive 2008/56/EC of the European Parliament and of the Coucil of 17 June 2008, establishing a framework for community action in the field of marine environmental policy (Marine Strategy Framework Directive), Official Journal of the European Communities, 2008 Search PubMed.
- OSPAR Commission, Quality Status Report 2010, London, 2010 Search PubMed.
- J. Rogowska, E. Olkowska, W. Ratajczyk and L. Wolska, Environ. Toxicol. Chem., 2018, 37, 1523–1534 CrossRef CAS PubMed.
- P. Nuss and G. A. Blengini, Sci. Total Environ., 2018, 613–614, 569–578 CrossRef CAS PubMed.
- J. Wojcieszek, J. Szpunar and R. Lobinski, TrAC, Trends Anal. Chem., 2018, 104, 42–53 CrossRef CAS.
- J. Petersen, D. Pröfrock, A. Paschke, J. A. C. Broekaert and A. Prange, Environ. Sci.: Water Res. Technol., 2016, 2, 146–153 RSC.
- U. Förstner, in Chemical Methods for Assessing Bio-Available Metals in Sludges and Soils, ed. R. Lescher, Elsevier, London and New York, 1985, pp. 1–30 Search PubMed.
- F. Ackermann, H. Bergmann and U. Schleichert, Environ. Technol. Lett., 1983, 4, 317–328 CrossRef CAS.
- J. Sastre, A. Sahuquillo, M. Vidal and G. Rauret, Anal. Chim. Acta, 2002, 462, 59–72 CrossRef CAS.
- D. E. Güven and G. Akinci, Gazi Univ. J. Sci., 2011, 24, 29–34 Search PubMed.
- V. Chand and S. Prasad, Microchem. J., 2013, 111, 53–61 CrossRef CAS.
- J. Ščančar, R. Milačič and M. Horvat, Water, Air, Soil Pollut., 2000, 118, 87–99 CrossRef.
- A. J. de Groot, K. H. Zschuppel and W. Salomons, Hydrobiologia, 1982, 91, 689–695 CrossRef.
- S. Uchida, K. Tagami and K. Tabei, Anal. Chim. Acta, 2005, 535, 317–323 CrossRef CAS.
- J. Kaakinen, T. Kuokkanen, J. Pesonen and I. Välimäki, Soil Sediment Contam., 2014, 23, 437–451 CrossRef CAS.
- R. G. Silva, M. N. Nadagouda, J. Webster, S. Govindaswamy, K. D. Hristovski, R. G. Ford, C. L. Patterson and C. A. Impellitteri, Environ. Sci.: Processes Impacts, 2013, 15, 645–652 RSC.
- J. Yoshinaga, A. Nakama and K. Takata, Analyst, 1999, 124, 257–261 RSC.
- J. Begerow and L. Dunemann, in Sampling and Sample Preparation: Practical Guide for Analytical Chemists, ed. M. Stoeppler, Springer, Berlin, Heidelberg, 1997, pp. 155–166 Search PubMed.
- C. D. B. Amaral, L. L. Fialho, F. P. R. Camargo, C. Pirola and J. A. Nóbrega, Talanta, 2016, 160, 354–359 CrossRef CAS PubMed.
- D. H. Loring and R. T. T. Rantala, Earth-Sci. Rev., 1992, 32, 235–283 CrossRef CAS.
- S. K. Gupta, M. K. Vollmer and R. Krebs, Sci. Total Environ., 1996, 178, 11–20 CrossRef CAS.
- D. Bossert, D. A. Urban, M. Maceroni, L. Ackermann-Hirschi, L. Haeni, P. Yajan, M. Spuch-Calvar, B. Rothen-Rutishauser, L. Rodriguez-Lorenzo, A. Petri-Fink and F. Schwab, Sci. Rep., 2019, 9, 7938 CrossRef PubMed.
- A. Durand, Z. Chase, A. T. Townsend, T. Noble, E. Panietz and K. Goemann, Int. J. Environ. Anal. Chem., 2016, 96, 119–136 CrossRef CAS.
- M. Krachler, C. Mohl, H. Emons and W. Shotyk, J. Anal. At. Spectrom., 2002, 17, 844–851 RSC.
- Z. Hu, W. Zhang, Y. Liu, H. Chen, R. M. Gaschnig, K. Zong, M. Li, S. Gao and S. Hu, Chem. Geol., 2013, 355, 144–152 CrossRef CAS.
- T. T. Magaldi, M. S. Navarro and J. Enzweiler, Geostand. Geoanal. Res., 2019, 43, 189–208 CrossRef CAS.
- A. L. Bezerra de Oliveira, J. C. Afonso, L. da Silva, A. Alcover Neto, M. Castro Carneiro, L. I. Dias da Silva and M. I. Couto Monteiro, Geostand. Geoanal. Res., 2019, 43, 689–699 CrossRef CAS.
- I. G. Ryss, The chemistry of fluorine and its inorganic compounds, United States Atomic Energy Commission, Technical Information Service, Available from the National Technical Information Service, Oak Ridge, Tenn., Springfield, Va., 1960 Search PubMed.
- M. Krachler, C. Mohl, H. Emons and W. Shotyk, Spectrochim. Acta, Part B, 2002, 57, 1277–1289 CrossRef.
- A. Sapkota, M. Krachler, C. Scholz, A. K. Cheburkin and W. Shotyk, Anal. Chim. Acta, 2005, 540, 247–256 CrossRef CAS.
- B. Bicalho, I. Grant-Weaver, C. Sinn, M. W. Donner, S. Woodland, G. Pearson, S. Larter, J. Duke and W. Shotyk, Fuel, 2017, 206, 248–257 CrossRef CAS.
- L. Balcaen, E. Bolea-Fernandez, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2015, 894, 7–19 CrossRef CAS PubMed.
- E. Bolea-Fernandez, L. Balcaen, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2017, 32, 1660–1679 RSC.
- D. MacDougall and W. B. Crummett, et al. , Anal. Chem., 1980, 52, 2242–2249 CrossRef CAS.
- S. L. R. Ellison and A. Williams, Eurachem Guide: Quantifying Uncertainty in Analytical Measurement, 3 edn, 2012 Search PubMed.
- S. L. R. Ellison, B. Magnusson and U. Örnemark, Eurachem Guide: Template for Eurachem Guides – A Guide for Guide Editors, 1 edn, 2015 Search PubMed.
- Analytical Methods Committee, Anal. Methods, 2016, 8, 5553–5555 RSC.
- H. Takei, T. Yokoyama, A. Makishima and E. Nakamura, Proc. Jpn. Acad., Ser. B, 2001, 77, 13–17 CrossRef.
- E. I. Müller, M. F. Mesko, D. P. Moraes, M. d. G. A. Korn and É. M. M. Flores, in Microwave-Assisted Sample Preparation for Trace Element Analysis, ed. É. M. d. M. Flores, Elsevier, Amsterdam, 2014, pp. 99–142 Search PubMed.
- V. Roje, J. Braz. Chem. Soc., 2011, 22, 532–539 CrossRef CAS.
- Ž. Fiket, N. Mikac and G. Kniewald, Geostand. Geoanal. Res., 2017, 41, 123–135 CrossRef.
- A. Retzmann, T. Zimmermann, D. Pröfrock, T. Prohaska and J. Irrgeher, Anal. Bioanal. Chem., 2017, 409, 5463–5480 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ay01049a |
| This journal is © The Royal Society of Chemistry 2020 |