
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modification of polyacrylate sorbent coatings with carbodiimide crosslinker chemistry for sequence-selective DNA extraction using solid-phase microextraction†
Marcelino
Varona
a,
Olga I.
Shimelis
b and
Jared L.
Anderson
 *a
*a
aDepartment of Chemistry, Iowa State University, 1605 Gilman Hall, Ames, Iowa 50011, USA. E-mail: andersoj@iastate.edu
bMilliporeSigma, Bellefonte, Pennsylvania 16823, USA
First published on 9th June 2020
Abstract
Selective DNA extraction is immensely useful for the isolation and detection of low-abundance sequences. Oligonucleotide-modified substrates are often used to capture sequences of interest for downstream analysis. In this study, we explore the chemical modification of commercial-available polyacrylate solid-phase microextraction fibers for selective DNA analysis using carbodiimide crosslinker chemistry. Reproducible modification conditions are found and the fibers were subsequently applied for selective DNA analysis. Several experimental parameters such as stir-rate, desorption time, and buffer-type are optimized. The developed method was able to selectively extract the target DNA sequence (260 bp) in the presence of 100-fold excess interfering salmon testes DNA.
Introduction
Nucleic acids are essential biopolymers responsible for the storage, transfer, and regulation of genetic information within biological systems. In addition, nucleic acids represent valuable diagnostic molecules for the detection and identification of diseases.1,2 Biomolecular techniques such as quantitative polymerase chain reaction (qPCR) can provide detailed information regarding nucleic acid sequences present within a sample. However, these methods use highly sensitive enzymes that are susceptible to inhibition by molecules native to biological matrices.3 Therefore, to enable successful analysis of these essential biomarkers, they must first be isolated in sufficient quantity and purity.Total nucleic acid extraction methods typically rely on adsorption to silica particles4 or liquid–liquid extraction (phenol/chloroform extraction).5 While these methods can isolate large amounts of nucleic acid, they fall short in applications targeting specific and/or low-copy number sequences. In circulating tumor DNA analysis, these valuable sequences often comprise a small percentage of the total nucleic acid present (<1%) within a sample.6 Moreover, traditional detection methods such as qPCR can suffer from amplification bias, where the most abundant sequence is preferentially amplified leading to false negatives and inconsistent quantification.7 These issues can be overcome through the use of digital PCR,8 which is expensive and not easily accessible, or through the upstream enrichment of target sequences.9,10
Methods for the isolation of specific sequences leverage the natural ability of nucleic acids to recognize complementary sequences through Watson–Crick base pairing interactions. A popular platform for this process is performed using biotin-modified oligonucleotides.11 These probes can hybridize to their complementary sequence and be subsequently enriched using streptavidin-coated magnetic beads. However, beads are expensive, notoriously prone to aggregation,12,13 and require an external magnetic field for their recovery.
An alternative preconcentration technique to magnetic beads is solid-phase microextraction (SPME).14 Several studies have utilized SPME for the isolation of DNA and RNA from biological matrices.15,16 In particular, Nacham et al. demonstrated the ability to use carbodiimide coupling chemistry to functionalize commercially-available polyacrylate fibers (PA) with amine-functionalized oligodeoxythymine 20 (dT20) to develop a selective sorbent for mRNA extraction.17 However, it was observed that significant fiber-to-fiber differences existed when the modification chemistry was performed. The fibers were found to contain between 20 and 40 ng of oligo dT20 following the modification procedure.17
In this study, we optimize the coupling chemistry in order to increase the fiber-to-fiber reproducibility of modified PA fibers. We also apply for the first time modified PA fibers for the selective extraction of DNA. Experimental parameters such as stir speed, buffer composition, and desorption time were optimized. The modified PA fibers were found to selectively extract the target DNA sequence while maintaining selectivity in the presence of 100-fold excess interfering DNA.
The coupling reaction and quantification assay were performed as previously described.17 Further experimental details can be found in the ESI.† Conditions and a representative illustration of the quantification assay are shown in Fig. 1. In brief, a dual-labeled oligonucleotide containing an amine group and a fluorescein (FAM) fluorophore was reacted with the PA fibers, washed multiple times with deionized water to remove unreacted oligo, and subjected to DNase I treatment. The resulting solution was analyzed with a plate reader and the amount of fluorophore in free solution quantified using an external calibration curve (Fig. S1†). All DNA sequences used in this study can be found in Table S1 within the ESI.†
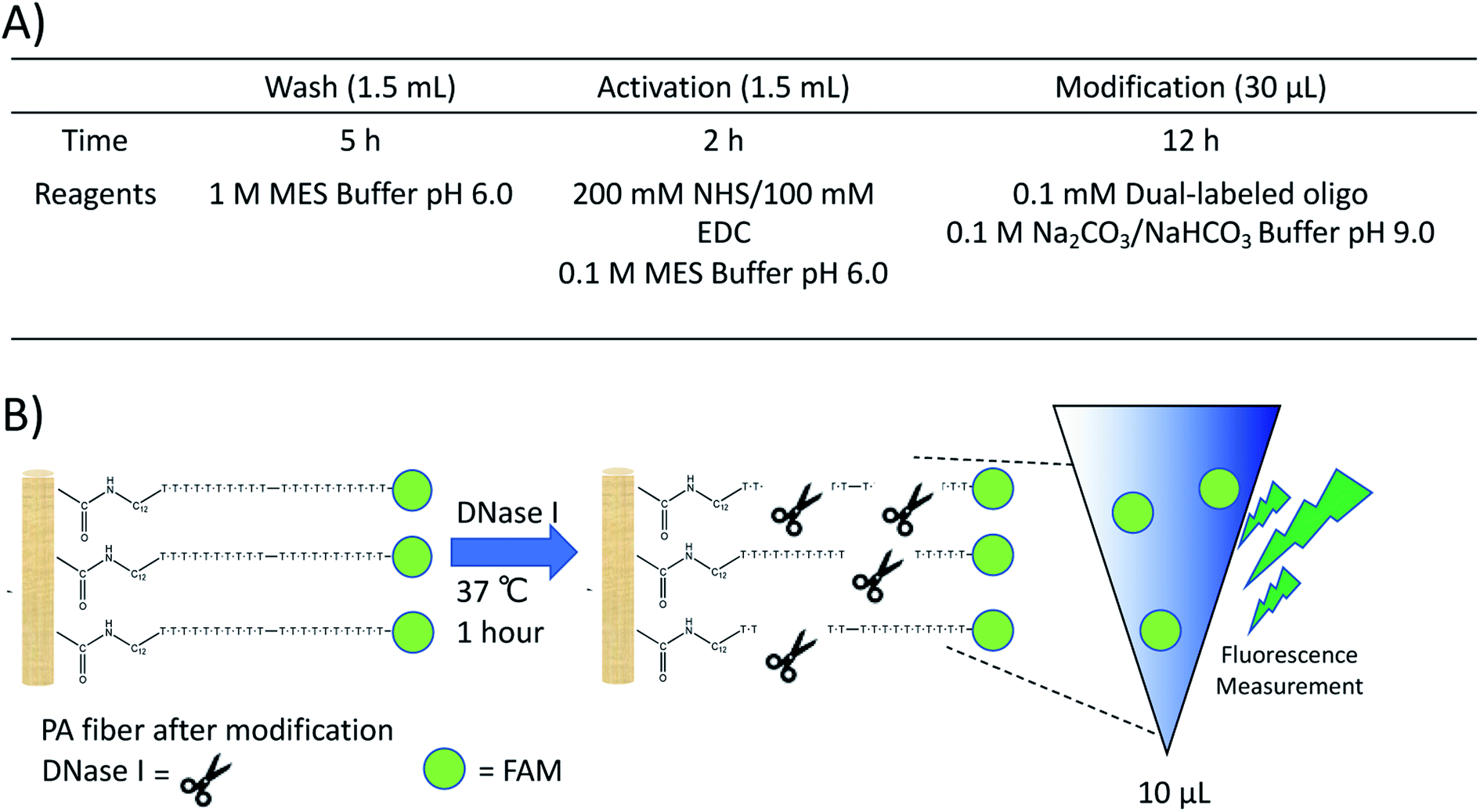 | ||
| Fig. 1 (A) Reaction conditions for the NHS/EDC modification of PA fibers and (B) representative schematic of the DNase assay used for quantification. | ||
Results and discussion
Initially, the effect of conditioning fibers at 280 °C for 30 min on the loading of the oligo was explored. Two previously conditioned and two unconditioned fibers were subjected to carbodiimide crosslinker chemistry, multiple wash steps to remove unreacted oligo, and then the DNase assay. Table S2† shows that multiple washes over a 24 h period of time were required to remove residual oligo not bound to the fibers. Results showing the mass of bound DNA on the fibers obtained after modification and DNase treatment are shown in Table 1. These results indicate that fiber conditioning did not improve the reproducibility of the modification. It was previously hypothesized that the loading efficiency was inconsistent due to lot-to-lot variation in the PA fibers.17 This variation could result in a different number of acid groups available for the coupling reaction.To test the reproducibility of the coupling reaction and DNase assay, a well-characterized support containing carboxylic acid groups (Supelco DSC-WCX ion exchange resin) was used. The reaction was performed as previously described on 1.2 mg of the particles. Following the DNase assay and fluorescence quantification, a total of 98.5 ng of oligo was able to be loaded onto the particles. Several more reactions were performed under the same conditions and the loading efficiency was found to have a relative standard deviation (RSD) > 23% (Table 2). This result indicated that reproducibility issues could be due to the employed reaction conditions. In particular, the pH of the coupling solution (pH 9) may affect reproducibility, as previous reports have utilized 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 6) in all steps. Once the coupling reaction was performed in MES buffer, reproducibility was observed to increase substantially. The RSD of triplicate reactions performed in MES dropped significantly to 1.2% (Table 2). These results indicated that the reaction must be performed under the appropriate conditions in order to achieve high reproducibility.
| Buffer in coupling step | Mass (n = 3) | RSD (n = 3) |
|---|---|---|
| Carbonate buffer | 89.11 | 23.44 |
| MES | 110.36 | 1.25 |
Using the previously optimized conditions, reactions were performed on six PA fibers from two different lots, as indicated by the manufacturer. Fiber-to-fiber reproducibility was tested by performing extractions of a 260 bp model sequence, as illustrated in Fig. 2. The extraction performance was monitored using quantitative polymerase chain reaction (qPCR) which amplifies the target DNA exponentially. An external calibration curve was prepared using the target DNA (Fig. S2†) and was found to have an amplification efficiency of 102.31%, within the acceptable limits set by the MIQE guidelines.18 For reference, a quantification cycle (Cq) value difference of one is equal to a two-fold difference in the amount of DNA present. Higher Cq values indicate lower amounts of DNA. Fig. S3† demonstrates the results obtained following DNA extractions and reveal consistent extraction performance within the same lot. However, there was lot-to-lot variability observed, as previously shown.17
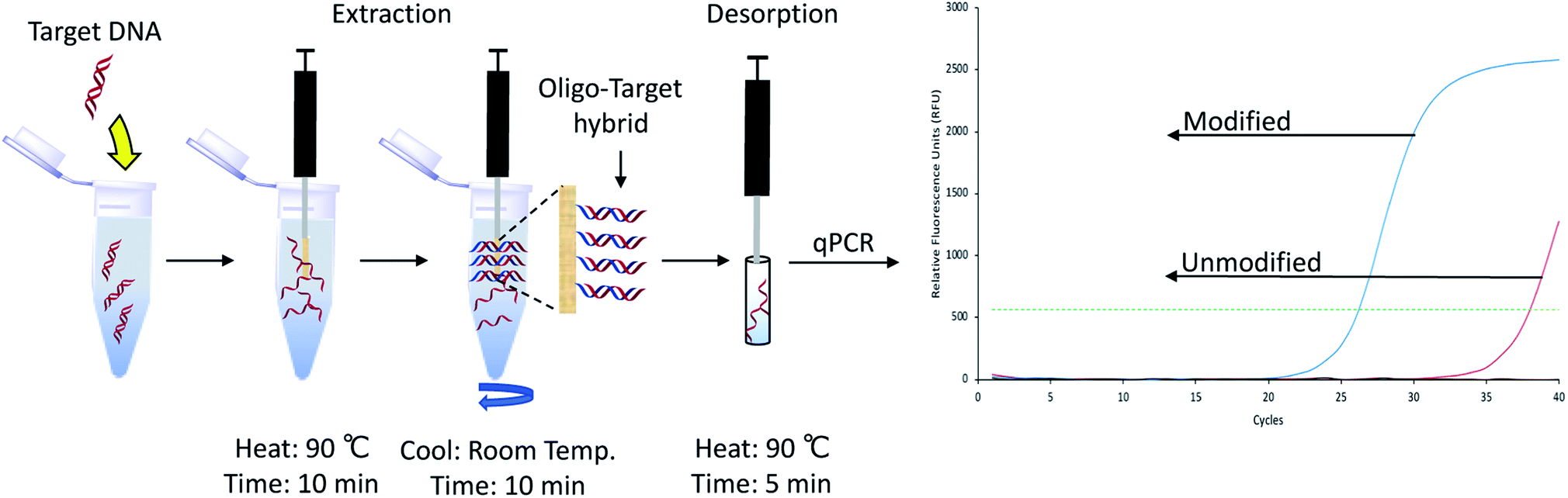 | ||
| Fig. 2 Representative schematic of the selective SPME process coupled to qPCR. | ||
One important aspect of the method workflow is the desorption step, as it is necessary to maximize the amount of DNA recovered and to also reduce carryover effects. To determine the optimal desorption time, serial desorptions were performed in 10 μL of water in different time intervals over a period of 20 min. A shown in Fig. 3A, 95% of the extracted DNA could be desorbed after 10 minutes, with 99% desorption efficiency being attained by 15 min. However, some DNA could still be detected by qPCR after 20 min (213 copies of target DNA).
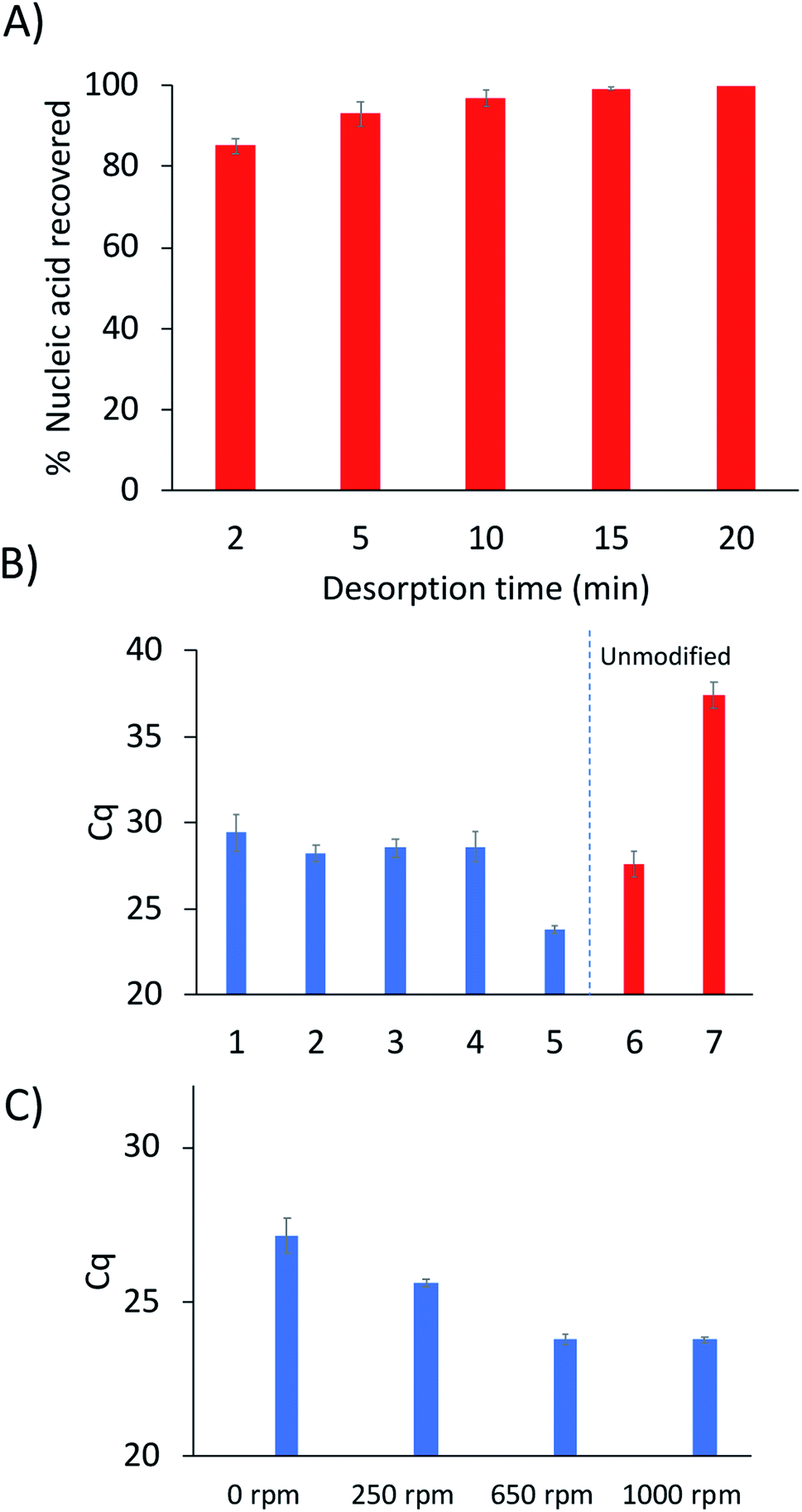 | ||
| Fig. 3 (A) Desorption time analysis following sequence-specific DNA extractions showing the percent DNA recovered during each time period. (B) Effect of buffer composition on the DNA extraction efficiency (1: 25 mM NaCl, 2 and 7: 250 mM NaCl, 3: 20 mM Tris pH 8.0, 4: 20 mM phosphate buffer pH 8.0, 5 and 6: citric acid–phosphate buffer pH 6). (C) Optimization of stir-rate during the annealing step on the extraction of DNA. For reference, a decrease of 1 in Cq value indicates a doubling of the DNA present in the qPCR reaction. | ||
It was hypothesized that the use of a nuclease could permit the removal of undesorbed DNA as a means to prevent carryover. To test this, exonuclease III was chosen to selectively remove the remaining DNA from the fiber. To prevent hydrolyzation of the probe, a spacer was added to the 3′ end to act as a protecting group. Fig. S4† shows a schematic that demonstrates the proposed mechanism of action of the enzyme within the system. The process was tested by performing an extraction and an initial desorption followed by treatment with exonuclease III and a subsequent desorption step. This result was compared with performing the second desorption without any previous enzyme treatment. Table S3† shows the ratio of DNA in the first desorption to the second desorption. These results show that treating the fibers with exonuclease III was able to significantly decrease carryover.
Nucleic acid hybridization has been shown to be highly dependent on the surrounding environment.19 Therefore, the buffer composition of the sample solution would be expected to play a role in the selective extraction of the target sequence. Extractions were performed from five different buffers to determine the composition that yielded the highest capture of target sequence (Fig. 3B). As expected, the 250 mM NaCl solution yielded higher extraction of DNA compared to the 25 mM NaCl solution. This is due to higher DNA-duplex stability at increased ionic strength, resulting in higher melting temperatures of the probe-target complex.19 In contrast, the Tris and phosphate buffers did not yield different extraction results from each other or from the 25 mM NaCl solution. However, a disadvantage to using phosphate buffers is the chelation of magnesium by the phosphate groups which can cause PCR inhibition.20
Interestingly, the extractions from citric acid–phosphate buffer yielded much higher quantity of captured DNA compared to the 250 mM NaCl solution (Fig. 3B). Extractions from the citric acid–phosphate buffer were approximately 16-fold higher than those from the NaCl solution. However, when extractions were performed with an unmodified fiber from the citric acid–phosphate buffer, a significant amount of DNA was still detected (approximately 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 copies). In contrast, the unmodified fiber in 250 mM NaCl extracted ≈18 copies, indicating 1000-fold lower non-specific DNA extraction than in citric acid–phosphate buffer. These results will be further explored in future studies, as previous work showed very low nonspecific extraction of DNA by PA fibers when extractions were performed from a Tris buffer.
000 copies). In contrast, the unmodified fiber in 250 mM NaCl extracted ≈18 copies, indicating 1000-fold lower non-specific DNA extraction than in citric acid–phosphate buffer. These results will be further explored in future studies, as previous work showed very low nonspecific extraction of DNA by PA fibers when extractions were performed from a Tris buffer.
The stir-rate was the final parameter optimized. In traditional SPME, the analysis time can often be decreased through the use of agitation. However, stirring had not previously been explored using hybridization-based SPME. In order to evaluate the effect of the stir-rate on the extraction of DNA, stirring was introduced into the hybridization step and varied between 0–1000 rpm. As shown in Fig. 3C, the amount of captured DNA can be observed to increase from 0–650 rpm. No difference in the amount of DNA extracted was observed when the agitation speed was increased from 650 to 1000 rpm.
Sequence-specific nucleic acid extraction methods are highly desirable when non-target DNA is present in large amounts relative to the target sequence. Therefore, these methods must possess high enough selectivity to isolate the target when interfering sequences are present. As a proof-of-concept, extractions were performed as previously described with salmon testes DNA (average length = 2000 base pairs) present as the interfering sequence at a concentration of 10 ng mL−1. This concentration is 100 times higher than the target DNA concentration (10 pg mL−1). The extraction results in Fig. 4 show little difference in the amount of DNA extracted when the salmon testes DNA is present and compared to an extraction without interfering DNA. These results demonstrate the high selectivity of the developed method.
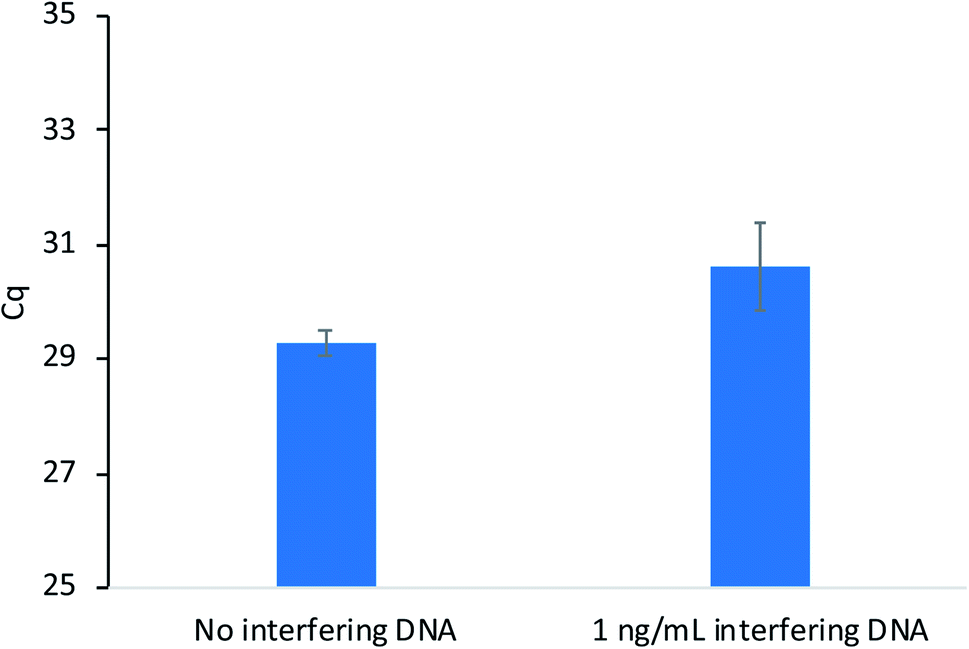 | ||
| Fig. 4 Extraction performance of modified PA fibers with and without interfering salmon testes DNA present. | ||
Conclusions
In conclusion, the modification of PA fibers and their application for selective DNA analysis was explored. Reproducible NHS/EDC reaction conditions were studied in order to decrease the fiber-to-fiber variability. Carryover DNA from previous extractions was able to be minimized using exonuclease III after the extraction procedure. A blocking group was added to the DNA probe bound to the fiber to prevent degradation by the exonuclease. The optimal extraction buffer and stir speed were also determined. Extractions of the target DNA were able to be performed in 100-fold excess interfering DNA. Subsequent studies will focus on further studying the selectivity of the sorbents for the extraction of single-nucleotide polymorphisms and the performance of the modified fibers in biological matrices. In addition, this study allows for reproducible modification of PA fibers with other bioactive molecules, such as proteins or antibodies, for the future development of selective sorbents.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge funding from the Chemical Measurement and Imaging Program at the National Science Foundation (Grant No. CHE-1709372).References
- E. Heitzer, P. Ulz and J. B. Geigl, Clin. Chem., 2015, 61, 112–123 CrossRef CAS PubMed.
- D. Helb, M. Jones, E. Story, C. Boehme, E. Wallace, K. Ho, J. Kop, M. R. Owens, R. Rodgers, P. Banada, H. Safi, R. Blakemore, N. T. N. Lan, E. C. Jones-López, M. Levi, M. Burday, I. Ayakaka, R. D. Mugerwa, B. McMillan, E. Winn-Deen, L. Christel, P. Dailey, M. D. Perkins, D. H. Persing and D. Alland, J. Clin. Microbiol., 2010, 48, 229–237 CrossRef CAS PubMed.
- P. Rådström, R. Knutsson, P. Wolffs, M. Lövenklev and C. Löfström, Appl. Biochem. Biotechnol., Part B Mol. Biotechnol., 2004, 26, 133–146 Search PubMed.
- R. Boom, C. J. A. Sol, M. M. M. Salimans, C. L. Jancen, P. M. E. Wertheim-van Dillen and J. van der Noordaa, J. Clin. Microbiol., 1990, 28, 495–503 CrossRef CAS.
- R. Patel, J. T. Kvach and P. Mounts, J. Gen. Microbiol., 1986, 132, 541–551 CAS.
- S. O. Kelley, ACS Sens., 2017, 2, 193–197 CrossRef CAS PubMed.
- P. M. Warnecke, C. Stirzaker, J. R. Melki, D. S. Millar, C. L. Paul and S. J. Clark, Nucleic Acids Res., 1997, 25, 4422–4426 CrossRef CAS PubMed.
- B. Vogelstein and K. W. Kinzler, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9236–9241 CrossRef CAS PubMed.
- M. Lovett, Curr. Protoc. Hum. Genet., 1994, 3–6 Search PubMed.
- A. B. Schrock, D. Pavlick, S. J. Klempner, J. H. Chung, B. Forcier, A. Welsh, L. Young, B. Leyland-Jones, R. Bordoni, R. D. Carvajal, J. Chao, R. Kurzrock, J. K. Sicklick, J. S. Ross, P. J. Stephens, C. Devoe, F. Braiteh, S. M. Ali and V. A. Miller, Clin. Cancer Res., 2018, 24, 1881–1890 CrossRef CAS PubMed.
- A. Holmberg, A. Blomstergren, O. Nord, M. Lukacs, J. Lundeberg and M. Uhlén, Electrophoresis, 2005, 26, 501–510 CrossRef CAS PubMed.
- D. C. Leslie, J. Li, B. C. Strachan, M. R. Begley, D. Finkler, L. A. L. Bazydlo, N. S. Barker, D. M. Haverstick, M. Utz and J. P. Landers, J. Am. Chem. Soc., 2012, 134, 5689–5696 CrossRef CAS PubMed.
- D. Liu, G. Liang, Q. Zhang and B. Chen, Anal. Chem., 2013, 85, 4698–4704 CrossRef CAS PubMed.
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145–2148 CrossRef CAS.
- O. Nacham, K. D. Clark and J. L. Anderson, Anal. Chem., 2016, 88, 7813–7820 CrossRef CAS PubMed.
- M. Varona, X. Ding, K. D. Clark and J. L. Anderson, Anal. Chem., 2018, 90, 6922–6928 CrossRef CAS PubMed.
- O. Nacham, K. D. Clark, M. Varona and J. L. Anderson, Anal. Chem., 2017, 89, 10661–10666 CrossRef CAS PubMed.
- J. F. Huggett, C. A. Foy, V. Benes, K. Emslie, J. A. Garson, R. Haynes, J. Hellemans, M. Kubista, R. D. Mueller, T. Nolan, M. W. Pfaffl, G. L. Shipley, J. Vandesompele, C. T. Wittwer and S. A. Bustin, Clin. Chem., 2013, 59, 892–902 CrossRef CAS PubMed.
- J. Fuchs, J. B. Fiche, A. Buhot, R. Calemczuk and T. Livache, Biophys. J., 2010, 99, 1886–1895 CrossRef CAS PubMed.
- S. R. Johnson, D. H. Martin, C. Cammarata and S. A. Morse, J. Clin. Microbiol., 1995, 33, 1036–1038 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ay00980f |
| This journal is © The Royal Society of Chemistry 2020 |