
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A de-waxing methodology for scanning probe microscopy
Safaa
Al Jedani
a,
Caroline I.
Smith
 a,
Philip
Gunning
b,
Barnaby G.
Ellis
a,
Peter
Gardner
c,
Steve D.
Barrett
a,
Asterios
Triantafyllou
d,
Janet M.
Risk
b and
Peter
Weightman
*a
a,
Philip
Gunning
b,
Barnaby G.
Ellis
a,
Peter
Gardner
c,
Steve D.
Barrett
a,
Asterios
Triantafyllou
d,
Janet M.
Risk
b and
Peter
Weightman
*a
aDepartment of Physics, University of Liverpool, L69 7ZE, UK. E-mail: peterw@liverpool.ac.uk
bDepartment of Molecular and Clinical Cancer Medicine, Institute of Translational Medicine, University of Liverpool, L3 9TA, UK
cManchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK
dDepartment of Pathology, Liverpool Clinical Laboratories, University of Liverpool, Liverpool, UK
First published on 16th June 2020
Abstract
A de-waxing protocol that successfully removes paraffin from tissue microarray (TMA) cores of fixed tissue obtained from oral cancer is described. The success of the protocol is demonstrated by the comparison of Fourier transform infrared (FTIR) results obtained on paraffin-embedded and de-waxed tissue and the absence of any significant correlations between infrared scanning near-field optical microscopy (SNOM) images of de-waxed tissue obtained at the three main paraffin IR peaks. The success of the protocol in removing paraffin from tissue is also demonstrated by images obtained with scanning electron microscopy (SEM) and by energy dispersive spectra (EDS) of a de-waxed CaF2 disc which shows no significant contribution from carbon. The FTIR spectra of the de-waxed TMA core overlaps that obtained from OE19 oesophageal cancer cells which had never been exposed to paraffin.
Introduction
There has been extensive research on the characterisation of cancerous cells and tissue using a variety of infrared (IR) imaging and spectroscopy techniques.1–5 Many of these studies involve experiments on tissue obtained in biopsies that have been formalin fixed and embedded in paraffin. Unfortunately, paraffin contributes to IR signals in regions of C–H stretching (3000–2800 cm−1) and C–H bending (1500–1350 cm−1) vibrations2 and these bands may mask some important contributions to the IR spectrum of the specimen such as those arising from lipids. One approach is to perform IR experiments on paraffin-embedded specimens and either remove the spectral contributions from paraffin digitally6,7 or remove these contributions from the analysis since the remaining portions of the spectrum contain significant information for spectral histopathology classification purposes. Alternatively, the paraffin can be removed from the tissue by a variety of “de-waxing” procedures in order to remove the paraffin bands from the IR spectrum. There is a wide variety of de-waxing protocols that can be used to remove wax from tissue. For pathological assessment of tissues, the method should be quick, easy and be good enough that the specific histochemical stain or stains required will be taken up by the tissue such that the pathologist can make the necessary evaluation. Small amounts of residual wax would not be noticed by these visual inspections – so long as the stain works the dewaxing is deemed successful. The situation is slightly different for spectroscopic evaluation since small amounts of residual wax can greatly influence the spectra observed. It is important therefore that a protocol be established where there is no spectral interference from residual wax. There are, of course, other studies that have investigated this process for the purposes of FTIR evaluation of tissue. These use different solvents, for example xylene, hexane, Citroclear to mention just three8 or have compared the effects of submersion time of the tissue in the de-waxing agent, on the IR spectrum. The literature presents conflicting results on the efficacy of de-waxing procedures,9–12 with some studies showing that not all the paraffin has been removed and still contributes to the IR spectrum10 and it is not clear which solvent is more effective at removing paraffin, though hexane has the advantage that it is less toxic.10,11 Most previous studies have focused on the impact on the sample of longer immersion times, varying from sixteen to twenty hours, with mild stirring or shaking to improve the de-waxing outcome, in either xylene or hexane. However, longer immersion times heightened the risk of losing some of the specimen especially in the case of tissue microarrays (TMA). Some studies have explored the effect of de-waxing at temperatures of 40 °C or higher12–14 with varying immersion and shaking times, but safety considerations are paramount when utilising xylene at these temperatures that lie so far above its flash point. A common concern is that immersion in solvents will remove important native hydrocarbons such as lipids from the specimens and such processes are expected to be enhanced at higher temperatures. However a detailed study12 indicates that although immersion in solvents to remove paraffin will leach the methylene chains of free, unbound lipids from tissue, solvent-resistant lipids will remain in formalin-fixed tissue by being locked into protein–lipid complex matrices, predominantly in membranes. This means that since there are features in the IR spectrum which arise from both paraffin and lipids it is difficult to demonstrate from an IR spectrum that a de-waxing procedure has removed all the paraffin from a specimen.This work reports the results of investigations of the influence of temperature on de-waxing and describes a protocol that efficiently removes paraffin from tissue at a temperature of 25 °C. This is confirmed by Fourier transform infrared spectroscopy (FTIR), infrared scanning near-field optical microscopy (IR-SNOM) and scanning electron microscopy (SEM) of waxed and de-waxed specimens of oral cancer tissue. The development of an effective method of removing paraffin from tissue is particularly important for studies with the surface sensitive SNOM technique, where wax remaining on the surface of a specimen will more strongly affect the resulting spectrum than in FTIR. The SNOM technique is slower than FTIR since images are collected one wavelength at a time. However SNOM is not diffraction limited and has the potential to provide chemical images of structures such as chromosomes and microvesicles that play important roles in the development of disease.15 A protocol that works for the SNOM therefore needs to be established.
Experimental
The paraffin used in this study was Histoplast paraffin (Thermo Scientific, Paisley, UK) which has a melting point of 56 °C and the paraffin is supplied mixed with plastic polymers. A formalin-fixed paraffin-embedded (FFPE) TMA containing three, one-millimetre diameter tissue cores of oral cancer tissue was constructed for pilot experiments. Tissue was obtained from the local pathology stores after written, informed consent was obtained from patients under ethical approval numbers EC 47.01 and 10/H1002/53. Five μm thick sections were cut from the TMA and floated onto calcium fluoride (CaF2) discs of 20 mm diameter and thickness of 2 mm (Crystran Ltd, Poole, UK). These samples were then baked in an oven at 37 °C for 4 hours to evaporate any excess water. Sections of blank paraffin were prepared in similar way to the tissue.Prior to the experiments all tools were cleaned by washing in deionised water and then autoclaved. An extensive series of pilot experiments were performed to determine the optimum procedure for removing paraffin from biological tissue by controlling the immersion time in the solvent and the temperature of the solvent. The solvents used were histological grade xylene (98.5%) from Sigma-Aldrich and absolute ethanol. Xylene is highly toxic and flammable and should be used in a fume hood.
Each CaF2 disc was immersed in a beaker of xylene and sequentially transferred to a second and third beaker with each immersion accompanied by a gentle swirling of the xylene. The specimen was then immersed in a sequence of three beakers of absolute ethanol for 5 minutes each accompanied by a gentle swirling. The temperature (10 °C, 15 °C, 20 °C and 25 °C) and timing (5, 10, 15 and 20 minutes) of the xylene washes were altered between experiments. Identical pilot experiments were performed on thirty-two CaF2 discs supporting either TMA sections or paraffin only and repeated three times.
The outcome of these de-waxing experiments was monitored by viewing the infrared (IR) spectra using a Bruker attenuated total reflection (ATR)-FTIR alpha spectrometer equipped with robust pressure clamps to ensure contact between the samples and a diamond ATR crystal. All results were recorded in transmission mode over the range 400 to 4000 cm−1 with a spectral resolution of 2 cm−1 using Bruker OPUS spectroscopy software and were the average of 100 individual scans of both the specimen and the background to improve signal-to-noise. ATR spectra were collected from three different regions from every disc of paraffin and from two regions from within the tissue cores and one from the areas between the cores (i.e. containing only paraffin) for the TMA specimens. The ATR spectra were treated by applying the rubber band baseline correction, wavelet de-noised then finally vector normalised.
After establishing the optimum conditions for removing paraffin from tissue, additional IR imaging experiments were performed on a further 6-core oral cancer TMA sectioned onto two CaF2 discs and that were either untreated or treated using the optimum de-waxing procedure. These experiments were carried out on an Agilent Cary 670-FTIR spectrometer in conjunction with an Agilent Cary 620-FTIR imaging microscope produced by Varian with a 128 × 128 pixel mercury–cadmium–telluride (MCT) focal plane array with a pixel size of 5.5 μm. The spectra were corrected for atmospheric and substrate absorption and the efficiencies of individual pixels in the array. FTIR images were acquired with a spectral range from 990 cm−1 to 3800 cm−1 with a spectral resolution of 4 cm−1, co-adding 64 scans for the images and 128 scans to determine the background. FTIR spectra were corrected with resonant Mie scattering16–19 and then principal component analysis (PCA) noise reduction was used to improve the signal-to-noise with the first 15 PCs being retained.
Areas of the waxed and de-waxed TMA cores studied by FTIR were also imaged using the aperture IR-SNOM described previously.20 In these experiments the SNOM was equipped with a MIRCat quantum cascade laser (QCL) instrument (Daylight Solutions, San Diego, USA) operating with modules covering the wavelength range of 1965 cm−1 to 1145 cm−1 in pulse mode with a repetition rate of 100 kHz and a pulse duration of 40–500 ns. IR-SNOM images were obtained on the same waxed and de-waxed specimens used in the FTIR experiments at wavenumbers characteristic of paraffin; 1378 cm−1, 1462 cm−1 and 1473 cm−1 and at wavenumbers used in a recent SNOM study of a single cancerous cell, 1242 cm−1, 1369 cm−1, 1539 cm−1, 1650 cm−1, and 1751 cm−1.5 The IR-transmitting fiber used was a cleaved 6 μm core diameter chalcogenide fiber (CorActive, Quebec, Canada). The signal-to-noise in the SNOM experiments was assessed by comparing the images obtained with the tip moving in the forward and backwards directions. In these experiments the signal-to-noise was of the order of 100.
Waxed and de-waxed specimens were also studied with a JEOL JSM-6610 scanning electron microscope (SEM) coupled with Tungsten filament and Oxford INCA X-act energy dispersive X-ray detector. The tissue samples were coated with gold or chromium before imaging to reduce charging as they are insulating materials. SEM images were obtained from TMA cores supported on a CaF2 disc before and after the optimum de-waxing procedure using a 10–15 kV accelerating voltage and variable pressure ranging from 10 to 270 Pa.
Results and discussion
Preliminary experiments showed that increasing the number and duration of washes at room temperature did not totally remove paraffin from the tissue even after 24 or 48 hours in xylene and demonstrated remaining residue which was visible to the naked eye. However, variations in ambient temperature during the xylene washes were found to have a major influence on the removal of paraffin and this led to a series of controlled experiments in which the temperature of the xylene and the total immersion time were varied.Fig. 1 shows the ATR-FTIR spectra of a blank CaF2 disc coated in paraffin and four separate discs after 60 minutes (3 × 20 minutes) in xylene at 10 °C, 15 °C, 20 °C and 25 °C. The effects of varying the immersion times of 15, 30 and 45 minutes were also studied but were found to leave residue on the discs at all temperatures. In the experiments at 10 °C and 15 °C the paraffin tended to clump together and after processing at these temperatures there were peaks in the spectrum associated with paraffin (Fig. 1). The 20 °C wash left a small residual signal which indicates that most of the paraffin was removed but there was a visible stain left on the disc which may be due to the plastic polymers that are mixed in with paraffin. The 25 °C wash showed there was no visible residue and a very flat spectrum in the ATR (Fig. 1) that could not be distinguished from noise. Further ATR studies established that the optimum conditions for de-waxing are three 20 minute washes with fresh xylene at 25 °C followed by three 5 minute washes in absolute ethanol.
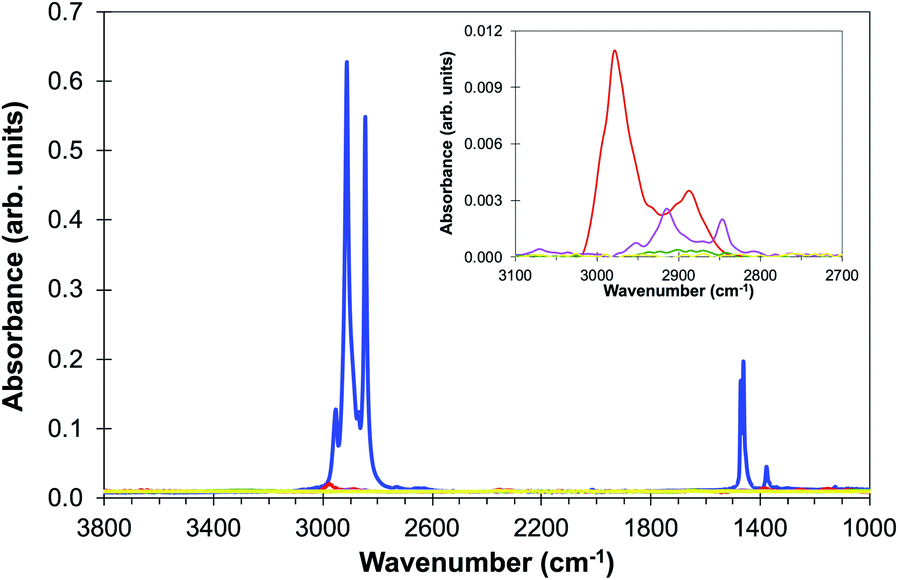 | ||
| Fig. 1 ATR spectra of (a) paraffin (blue line) and de-waxed discs after 60 minutes in xylene at (b) 10 °C (red line), (c) 15 °C (pink line), (d) 20 °C (green line) and (e) 25 °C (yellow line). The inset shows the most intense paraffin region of the spectrum in more detail. It is likely that the upper curve in the inset includes contributions from xylene since the peak extends to higher wavenumbers than the paraffin peaks. | ||
Experiments were then carried out on two sequential sections of the 6-core TMA: one specimen was imaged before any processing and one after the optimum de-waxing procedure described above. The average absorption spectra obtained from within the tissue core and from the area of the CaF2 disc outside the tissue using the FTIR imaging microscope with a focal plane array (FPA) are shown for both experiments in Fig. 2(a) and (b). Wavelengths attributable to the paraffin signal are very strongly absorbed in the waxed TMA core and the CaF2 disc while the de-waxing procedure leads to very low absorbance in both core and the CaF2 disc [Fig. 2(a) and (b)]. This does not however prove that there are not areas of residual paraffin in the TMA core. Fig. 2(c) shows a comparison of the FTIR spectra of paraffin, the TMA core embedded in wax and the de-waxed core. The paraffin peaks at 1378 cm−1, 1462 cm−1 and 1473 cm−1 can be clearly seen in the waxed tissue; weak features in the FTIR of the de-waxed tissue are visible in the same region.
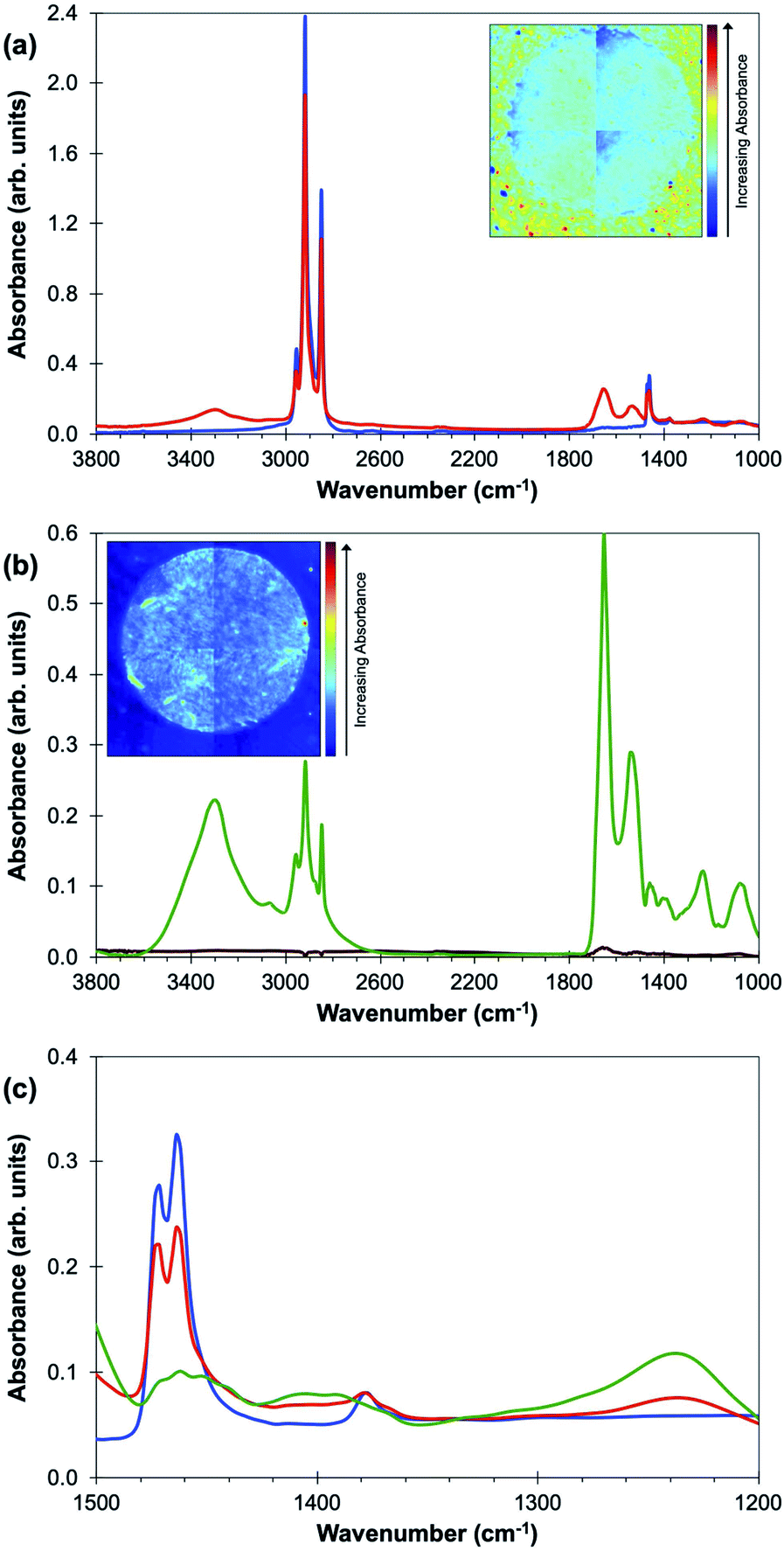 | ||
| Fig. 2 (a) Average FTIR absorbance spectra of a waxed tissue core (red line) and area of the CaF2 disc outside the tissue (blue line), (b) average FTIR absorbance spectra of a de-waxed tissue core (green line) and area of the CaF2 disc outside the tissue (brown line) and (c) is an expansion of the region between 1500 cm−1 and 1200 cm−1 for the FTIR spectra of paraffin, the waxed and de-waxed core. The two strong paraffin peaks are at 1473 cm−1 and 1462 cm−1 with a weaker peak at 1378 cm−1. The inseted images in both graphs show the FTIR absorbance intensity at 1462 cm−1. The spectra in (b) have been corrected for Mie-scattering (note the more sensitive vertical scale in (b)). | ||
The SNOM technique collects spectra through a sharp tip close to the surface of a specimen and is expected to be particularly sensitive to the presence of any surface paraffin. In order to confirm that this de-waxing protocol is suitable for SNOM studies the waxed and de-waxed TMAs used in the FTIR imaging studies were imaged in the SNOM at a range of wavenumbers including those of the principal paraffin peaks, 1378 cm−1, 1462 cm−1 and 1473 cm−1 and those used in a recent SNOM study of a single cancerous cell, 1242 cm−1, 1369 cm−1, 1539 cm−1, 1650 cm−1, and 1751 cm−1.5 All these images were taken in a common area of the core showing the edge of the core with the bare CaF2 disc on the right-hand side of the image.
The boundary between the core and the CaF2 slide cannot be discerned in the waxed specimen [Fig. 3(a)] obtained at one of the strong paraffin peaks, 1462 cm−1, although there are regions of the specimen in which more light is transmitted (red colour) through the tissue core (left of image), than through the CaF2 slide (right of image). The boundary between the two regions can be discerned in images of the de-waxed TMA core obtained at 1462 cm−1 [Fig. 3(b)] and at the second of the strong paraffin peaks, 1473 cm−1 [Fig. 3(c)]. The boundary is also more clearly seen in the images of the de-waxed core obtained at a number of other wavelengths (Fig. 4).
 | ||
| Fig. 3 (a) and (b) show the IR-SNOM images of a waxed and de-waxed TMA core, respectively, at 1462 cm−1 and (c) shows the IR-SNOM image of the same de-waxed core at 1473 cm−1. Tissue is located to the left of each image and CaF2 slide to the right and the images are all 300 μm × 300 μm. Note that the intensity scale on the right that is used for all SNOM images in this paper has red denoting higher transmission and blue lower transmission – this is opposite to that used for the FTIR images. The rms noise level in the SNOM images is 1%. | ||
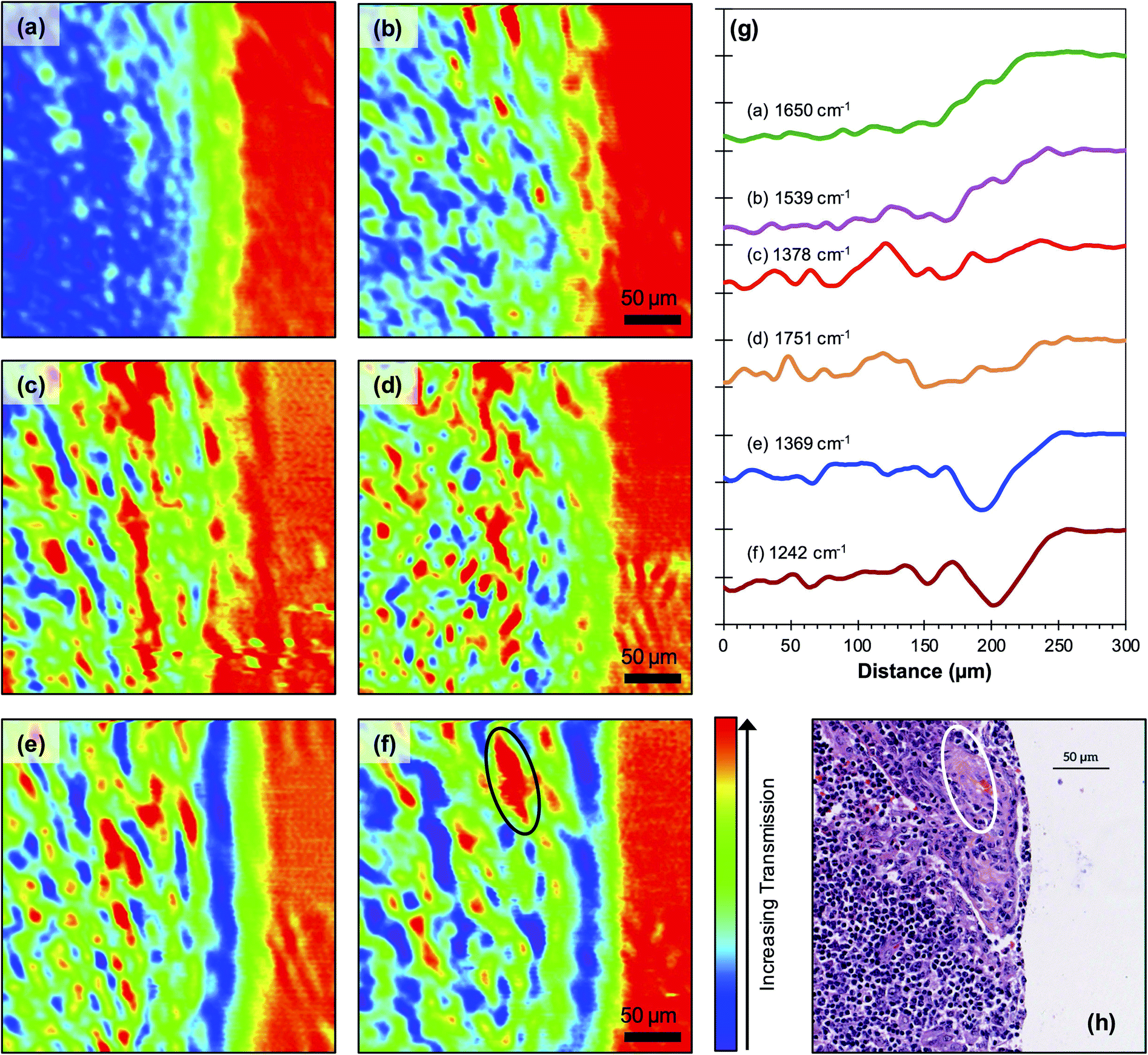 | ||
| Fig. 4 300 μm × 300 μm IR-SNOM images of a de-waxed TMA core at (a) 1650 cm−1, (b) 1539 cm−1, (c) 1378 cm−1, (d) 1751 cm−1, (e) 1369 cm−1 and (f) 1242 cm−1. Average line profiles over all images (a) to (f) are shown in (g). The corresponding H&E image is shown in (h). The area outlined in (f) and (h) shows a co-located region containing fewer nuclei. The rms noise level in the SNOM images is 1%. | ||
The images obtained of the waxed specimens at all wavelengths (not shown) show similar regions of increased transmission in the left-hand side of the image to those in Fig. 3(a). This indicates that the thick paraffin layer over the CaF2 slide is more effective at blocking transmission at all wavelengths than the combination of the core and the interspersed paraffin. Inspection of the images obtained at the three paraffin peaks 1462 cm−1, 1473 cm−1 [Fig. 3(b) and (c)] and 1378 cm−1 [Fig. 4(c)] shows that there is no clear correspondence in the location of the small regions of high transmission in the three images. In order to explore this observation in more detail a correlation analysis, described in an earlier paper,5 was performed on the images obtained of the de-waxed core at all the wavelengths studied (Table 1). If paraffin is still present in the tissue, one would expect a very high degree of correlation between the images obtained at the peaks in the paraffin spectrum. These correlations are very weak, 0.07 between the two strong peaks at 1462 cm−1 and 1473 cm−1 and 0.04 and 0.05 respectively with each of these peaks and the weak paraffin peak at 1378 cm−1 (Table 1). The correlation study together with the overlaps in the FTIR spectra shown in Fig. 2(c) confirm that the SNOM images obtained at 1462 cm−1, 1473 cm−1 and 1378 cm−1 do not arise from paraffin but from other contributors to the FTIR spectrum in these regions of the tissue. We conclude that there is no significant concentration of paraffin in the de-waxed core.
| Wavenumber (cm−1) | 1462 | 1650 | 1473 | 1378 | 1369 | 1242 | 1539 | 1751 |
|---|---|---|---|---|---|---|---|---|
| 1462 | 1.00 | −0.01 | 0.07 | 0.04 | 0.01 | 0.04 | −0.03 | −0.01 |
| 1650 | −0.01 | 1.00 | −0.11 | 0.19 | 0.09 | 0.22 | 0.30 | 0.27 |
| 1473 | 0.07 | −0.10 | 1.00 | 0.05 | −0.04 | 0.13 | 0.11 | −0.14 |
| 1378 | 0.04 | 0.19 | 0.05 | 1.00 | 0.06 | 0.18 | 0.43 | 0.45 |
| 1369 | 0.01 | 0.09 | −0.04 | 0.06 | 1.00 | 0.34 | 0.05 | 0.01 |
| 1242 | 0.04 | 0.22 | 0.13 | 0.18 | 0.34 | 1.00 | 0.16 | −0.03 |
| 1539 | −0.03 | 0.30 | 0.11 | 0.43 | 0.05 | 0.16 | 1.00 | 0.17 |
| 1751 | −0.01 | 0.27 | −0.18 | 0.45 | 0.01 | −0.03 | 0.17 | 1.00 |
The 1462 cm−1 peak is assigned to a bending (scissoring) vibration of methylene (CH2) groups in general and many biological molecules will also have methylene bending vibrations of similar strength. Consequently, the spectral contribution at 1462 cm−1 is not expected to completely disappear after de-waxing as indicated in earlier work21 on human prostate cells that have been formalin-fixed and not treated with paraffin. There are significant correlations between the images obtained at other wavenumbers and in particular with the image obtained at 1378 cm−1, which is weakly present in the paraffin spectrum. This latter one shows a correlation of 0.45 with the image obtained at 1751 cm−1, a wavenumber attributed to lipids, and of 0.43 with the image obtained at 1539 cm−1, the wavenumber of the Amide II band and which has been loosely attributed to β-sheets.5 The remaining significant correlation of 0.43 is between the images obtained at 1369 cm−1 and 1242 cm−1. Images at these two wavenumbers were also found to be correlated in the earlier study5 of a single cancer cell which is consistent with the attribution of the 1369 cm−1 and 1242 cm−1 wavenumbers respectively to the C–N stretch vibration of cytosine and guanine components of DNA22 and to the (PO2−) vibration of DNA.5
There are no sharp edges in any of the features in the SNOM images so it is not possible to determine an accurate value for the spatial resolution of the experiments but inspection of the images indicates that this is of the order of two to three pixels ∼5 μm ± 1 μm independent of wavelength. Individual features in the images obtained at all wavelengths shown in Fig. 4 show a similar distribution of sizes in the range 2 μm to 15 μm. The strong correlation between images obtained at particular wavelengths are also shown in the average intensity profiles across the core [Fig. 4(g)]. It is clear from these profiles that the chemical composition of the core changes significantly near the edge of the tissue. In particularly the intensities observed at all wavelengths rise significantly on approaching the edge. This probably reflects the fact that the profiles are averages over the whole of the images and there is some variation in the position of the edge of the core at different points in the image. In addition, the sharp edge of the tissue will be blurred by the finite spatial resolution of the SNOM. The line profiles also show significant correlations between the images obtained at (a) 1378 cm−1 [Fig. 4(c)] and 1751 cm−1 (lipids) [Fig. 4(d)] wavelengths suggesting that the contribution at 1378 cm−1 also arises from lipids, (b) the Amide I [1650 cm−1Fig. 4(a)] and Amide II [1539 cm−1Fig. 4(b)] wavelengths, which are representative of protein, and (c) the 1369 cm−1 [Fig. 4(e)] and 1242 cm−1 [Fig. 4(f)] wavelengths attributed to different constituents of DNA. The Haematoxylin and Eosin (H&E) image obtained from this region of the TMA core and co-located with the SNOM images to an accuracy of a few pixels is also shown (Fig. 4(h)). This shows that the majority of the cells in this region are lymphocytes with nuclei in the size range 4–8 μm, which is consistent with the size of features observed in the images obtained at the wavelengths associated with DNA. In particular, it should be noted that areas of high transmission at DNA-associated wavenumber 1242 cm−1 co-locate with likely keratinised areas demonstrating a lower density of DNA-containing nuclei (Fig. 4). The relationship between the H&E and 1242 cm−1 images is imperfect, and this can be attributed to the fact that the H&E and SNOM images were obtained from distinct, but adjacent 5 μm sections. Thus, due to the size of the individual cells, the distribution of the lymphocyte nuclei in the majority of the H&E image will be different to that in the 1242 cm−1 image, whereas the larger, cancer cell-containing structure in the top right of the images is retained in both sections.
A paraffin embedded TMA core was also studied with the SEM technique before and after de-waxing the specimen. Since the specimens are insulating, they were coated with a thin film of gold or chromium in the SEM experiments to reduce charging. Fig. 5(a) and (b) show respectively low magnification images (95×) of the waxed and de-waxed TMA core. The effect of removing the paraffin is marked, with only the edges of the TMA and folds in the tissue core distinguishable in Fig. 5(a) while Fig. 5(b) shows additional detail in the tissue of the dewaxed core. At higher magnification the image of the waxed specimen shows a swirly texture in the paraffin [Fig. 5(c)] which is missing in the image of the de-waxed sample [Fig. 5(d)] which shows a structure consistent with that expected of tissue. The efficiency of the de-waxing procedure was also investigated by collecting energy dispersive spectra (EDS) from areas of the CaF2 disc outside of a TMA core during the SEM experiments. This EDS signal was dominated by contributions from calcium and fluorine with only a negligible contribution from carbon demonstrating that the de-waxing procedure has removed the paraffin wax and also the thin Au coating deposited on the specimens for the SEM experiments.
 | ||
| Fig. 5 SEM images of (a) waxed TMA core, (b) de-waxed TMA core at low magnification and (c) waxed TMA core, (d) de-waxed TMA core at high magnification. | ||
A final demonstration that the FTIR spectra observed from the de-waxed core does arise from components of the tissue rather than from embedding paraffin is provided by the complete overlap of the FTIR spectrum of the de-waxed tissue core with that of formalin-fixed OE19 oesophageal cancer cells23 (Fig. 6) that have never been exposed to paraffin. Both spectral profiles show features in the 1440 cm−1 to 1480 cm−1 region of the spectra which are hidden by strong paraffin peaks in the waxed tissue [Fig. 2(c)].
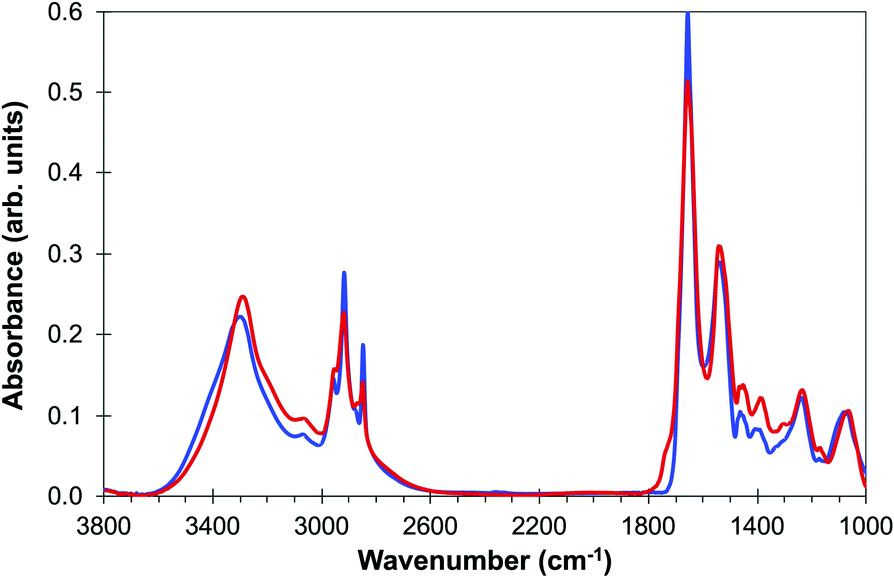 | ||
| Fig. 6 Comparison of average spectra of (a) de-waxed tissue core (blue line) and (b) OE19 oesophageal cell line (red line) adapted from ref. 23. | ||
Conclusions
We have demonstrated that the de-waxing protocol described above successfully removes paraffin from the TMA cores of fixed tissue studied in this work and is suitable for SNOM analysis. This conclusion is consistent with the data reported using the FTIR and confirmed by the absence of any significant correlations between the three paraffin peaks in the SNOM analysis, the results of the EDS spectrum of a de-waxed CaF2 disc which shows no significant contribution from carbon, and the overlap between the FTIR spectra of the de-waxed TMA core and that of the OE19 cells which had not been exposed to paraffin. The SNOM images obtained at a range of wavelengths shows the potential of the technique to yield detailed information on the chemical structure of tissue.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Cancer Research UK for funding (C7738/A26196). SAJ acknowledges the Saudi Arabia Scholarship Council for a Ph.D. studentship. BE acknowledges support from an EPSRC PhD studentship. The authors would like to thank Nathan Cumberbatch for help with the Bruker ATR. We would also like to thank the technical staff in the Central Teaching Laboratories, University of Liverpool for providing access to the ATR-FTIR. We also thank the Imaging Centre at Liverpool (ICaL) for providing access to the SEM and Dr Matthew Bilton for help with performing SEM imaging. We also acknowledge advice from Prof. A. Cricenti and Dr M. Luce of the Istituto di Struttura della Materia, CNR, Rome, on the operation of the SNOM.References
- T. P. Wrobel and R. Bhargava, Anal. Chem., 2018, 90, 1444 CrossRef CAS PubMed.
- M. Pilling and P. Gardner, Chem. Soc. Rev., 2016, 45, 1935 RSC.
- M. Diem, A. Mazur, K. Lenau, J. Schubert, B. Bird, M. Miljkovic, C. Krafft and J. Popp, J. Biophotonics, 2013, 6, 855 CrossRef CAS PubMed.
- I. P. Santos, E. M. Barroso, T. C. Bakker Schut, P. J. Caspers, C. G. F. van Lanschot, D.-H. Choi, M. F. van der Kamp, R. W. H. Smits, R. van Doorn, R. M. Verdijk, V. Noordhoek Hegt, J. H. von der Thüsen, C. H. M. van Deurzen, L. B. Koppert, G. J. L. H. van Leenders, P. C. Ewing-Graham, H. C. van Doorn, C. M. F. Dirven, M. B. Busstra, J. Hardillo, A. Sewnaik, I. ten Hove, H. Mast, D. A. Monserez, C. Meeuwis, T. Nijsten, E. B. Wolvius, R. J. Baatenburg de Jong, G. J. Puppels and S. Koljenović, Analyst, 2017, 142, 3025 RSC.
- J. Ingham, M. J. Pilling, T. Craig, M. R. F. Siggel-King, C. I. Smith, P. Gardner, A. Varro, D. M. Pritchard, S. D. Barrett, D. S. Martin, P. Harrison, P. Unsworth, J. D. Kumar, A. Wolski, A. Cricenti, M. Luce, M. Surman, Y. M. Saveliev and P. Weightman, Biomed. Phys. Eng. Express, 2018, 4, 025011 CrossRef.
- A. Tfayli, C. Gobinet, V. Vrable, R. Huez, M. Manfait and O. Piot, Appl. Spectrosc., 2009, 63, 564 CrossRef CAS PubMed.
- D. Sebiskveradze, C. Gobinet, E. Ly, M. Manfait, P. Jeannesson, M. Herbin, O. Piot and V. Vrabie, 8th IEEE International Conference on BioInformatics and BioEngineering, Athens, 2008, p. 1 Search PubMed.
- F. Lyng, E. Gazi, and P. Gardner, in Biomedical Applications of Synchrotron Radiation Infrared Microscopy, ed. D. Moss, Royal Society of Chemistry, Cambridge, 2010, ch. 5, pp. 145–191 Search PubMed.
- S. A. Mian, H. E. Colley, M. H. Thornhill and I. U. Rehman, Appl. Spectrosc. Rev., 2014, 49, 614 CrossRef CAS.
- J. Nallala, G. R. Lloyd and N. Stone, Analyst, 2015, 140, 2369 RSC.
- E. Ó. Faoláin, M. B. Hunter, J. M. Byrne, P. Kelehan, H. A. Lambkin, H. J. Byrne and F. M. Lyng, J. Histochem. Cytochem., 2005, 53, 121 CrossRef PubMed.
- C. Hughes, L. Gaunt, M. Brown, N. W. Clarke and P. Gardner, Anal. Methods, 2014, 6, 1028 RSC.
- D. C. Fernandez, R. Bhargava, S. M. Hewitt and I. W. Levin, Nat. Biotechnol., 2005, 23, 469 CrossRef CAS PubMed.
- J. D. Pallua, C. Pezzei, B. Zelger, G. Schaefer, I. K. Bittner, V. A. Huck-Pezzei, S. A. Schoenbichler, H. Hahn, A. Kloss-Brandstaetter, K. Kloss, G. K. Bonn and C. W. Huck, Analyst, 2012, 137, 3965 RSC.
- J. Ingham, T. Craig, C. I. Smith, A. Varro, D. M. Pritchard, S. D. Barrett, D. S. Martin, P. Harrison, P. Unsworth, J. D. Kumar, A. Wolski, A. Cricenti, M. Luce, M. Surman, Y. M. Saveliev, P. Weightman and M. R. F. Siggel-King, Biomed. Phys. Eng. Express, 2018, 5, 015009 CrossRef.
- P. Bassan, A. Kohler, H. Martens, J. Lee, H. J. Byrne, P. Dumas, E. Gazi, M. Brown, N. Clarke and P. Gardner, Analyst, 2010, 135, 268 RSC.
- H. Martens and E. Stark, J. Pharm. Biomed. Anal., 1991, 9, 625 CrossRef CAS PubMed.
- A. Kohler, J. Sulé-Suso, G. D. Sockalingum, M. Tobin, F. Bahrami, Y. Yang, J. Pijanka, P. Dumas, M. Cotte, D. G. van Pittius, G. Parkes and H. Martens, Appl. Spectrosc., 2008, 62, 259 CrossRef CAS PubMed.
- P. Bassan, H. J. Byrne, F. Bonnier, J. Lee, P. Dumas and P. Gardner, Analyst, 2009, 134, 1586 RSC.
- C. I. Smith, M. R. F. Siggel-King, J. Ingham, P. Harrison, D. S. Martin, A. Varro, D. M. Pritchard, M. Surman, S. Barrett and P. Weightman, Analyst, 2018, 143, 5912 RSC.
- M. J. Baker, C. Clarke, D. Démoulin, J. M. Nicholson, F. M. Lyng, H. J. Byrne, C. A. Hart, M. D. Brown, N. W. Clarke and P. Gardner, Analyst, 2010, 135, 887 RSC.
- A. C. S. Talari, M. A. Garcia Martinez, Z. Movasaghi, S. Rehman and I. Ur Rehman, Appl. Spectrosc. Rev., 2017, 52, 456 CrossRef CAS.
- J. Ingham, M. J. Pilling, D. S. Martin, C. I. Smith, B. G. Ellis, C. A. Whitley, M. R. F. Siggel-King, P. Harrison, T. Craig, A. Varro, D. M. Pritchard, A. Varga, P. Gardner, P. Weightman and S. Barrett, Infrared Phys. Technol., 2019, 102, 103007 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |