
A green liquid chromatography method for rapid determination of ergosterol in edible fungi based on matrix solid-phase dispersion extraction and a core–shell column†
 ab
ab
Abstract
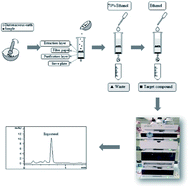
Developing a green analytical method for the analysis of components in food samples is an important research aspect of liquid chromatography (LC). The traditional LC method usually consumes a lot of toxic solvent for sample extraction and LC separation. In the current study, a green analytical method for the rapid determination of ergosterol in edible fungi was established. The sample was extracted and purified by matrix solid-phase dispersion (MSPD) with a green solution (ethanol and water). The LC separation was performed using a Poroshell 120 SB-C18 (4.6 × 30 mm, 2.7 μm) column with a green mobile phase (94% ethanol) at a flow rate of 1.0 mL min−1. The detection wavelength was set at 283 nm. The calibration curve of ergosterol showed good linearity (R = 0.9999) within the test range (4.21–25.27 μg mL−1). The RSD of precision was less than 2.0% and the recovery was 100.4% (RSD = 3.23%). The developed method was successfully applied to quantitative analysis of ergosterol in six edible fungi and the contents of ergosterol were in the range of 1.68–4.02 mg g−1. Only 11.5 mL ethanol water solution was used in the sample extraction and LC separation in the newly developed method, and no toxic organic solvents were used. The total analysis time was less than 15.5 min, about 12–14 min for sample extraction and 1.5 min for LC analysis. This method was environmentally friendly and time-saving, which is helpful to improve the quality evaluation of edible fungi.
 Please wait while we load your content...
Please wait while we load your content...

