
Analysis of intracellular α-keto acids by HPLC with fluorescence detection

Abstract
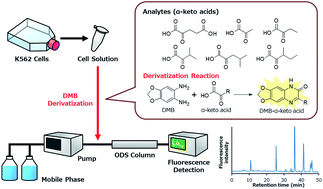
Branched-chain keto acids and branched-chain amino acids are metabolites of branched-chain amino acid aminotransferases (BCATs), which catalyzes reversible transamination between them. We found that BCAT1 plays an important role in the progression of myeloid leukaemia, and a method for the analysis of intracellular α-keto acids including branched-chain keto acids was necessary to further investigate their role. In this study, we developed a method to analyze six α-keto acids (α-ketoglutaric acid (KG), pyruvic acid, α-ketobutyric acid, α-ketoisovaleric acid, α-ketoisocaproic acid, and α-keto-β-methylvaleric acid) in K562 cells by HPLC with fluorescence detection, using 1,2-diamino-4,5-methylenedioxybenzene (DMB) as a derivatization reagent. Because split peaks of DMB-KG were observed when injection samples were too acidic, the derivatization solution was diluted with NaOH solution to obtain a single peak. Limits of detection and limits of quantification were 1.3–5.4 nM and 4.2–18 nM, respectively. Intracellular concentrations of α-keto acids were 1.55–316 pmol/1 × 106 K562 cells. The developed method realized reproducible and sensitive analysis of intracellular α-keto acids. Thus, the method could be used to elucidate the role of BCAT in myeloid leukaemia.
 Please wait while we load your content...
Please wait while we load your content...

