
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synchrotron radiation μ X-ray diffraction in transmission geometry for investigating the penetration depth of conservation treatments on cultural heritage stone materials
Elena
Possenti
 *a,
Claudia
Conti
a,
G. Diego
Gatta
b,
Marco
Merlini
b,
Marco
Realini
a and
Chiara
Colombo
a
*a,
Claudia
Conti
a,
G. Diego
Gatta
b,
Marco
Merlini
b,
Marco
Realini
a and
Chiara
Colombo
a
aIstituto di Scienze del Patrimonio Culturale (ISPC), Consiglio Nazionale delle Ricerche (CNR), Via R. Cozzi 53, 20125 Milano, Italy. E-mail: elena.possenti@cnr.it; claudia.conti@cnr.it; marco.realini@cnr.it; chiara.colombo@cnr.it; Tel: +39 02 66173386
bDipartimento di Scienze della Terra, Università degli Studi di Milano, Via S. Botticelli 23, 20133 Milano, Italy. E-mail: diego.gatta@unimi.it; marco.merlini@unimi.it
First published on 12th March 2020
Abstract
The assessment of the penetration depth of conservation treatments applied to cultural heritage stone materials is a burning issue in conservation science. Several analytical approaches have been proposed but, at present, many of them are not fully exhaustive to define in a direct way the composition and location of the conservation products formed after inorganic mineral treatments. Here, we explored, for the first time, the analytical capability of synchrotron radiation μ X-ray diffraction in transmission geometry (SR-μTXRD) for the study of the crystal chemistry and penetration depth of the consolidating phases formed after the application of diammonium hydrogen phosphate (DAP) treatments on a porous carbonatic stone (Noto limestone). The SR-μTXRD approach provided unambiguous information on the nature of the newly formed calcium phosphates (hydroxyapatite, HAP, and octacalcium phosphate, OCP) with depth, supplying important indications of the diffusion mechanism and the reactivity of the substrate. Qualitative and semi-quantitative data were obtained at the microscale with a non-destructive protocol and an outstanding signal-to-noise ratio. The SR-μTXRD approach opens a new analytical scenario for the investigation of a wide range of cultural heritage materials, including natural and artificial stone materials, painted stratigraphies, metals, glasses and their decay products. Furthermore, it can potentially be used to characterize the penetration depth of a phase “A” (or more crystalline phases) in a matrix “B” also beyond the cultural heritage field, demonstrating the potential wide impact of the study.
1 Introduction
Inorganic mineral treatments are applied for conservation purposes to decayed stone materials of cultural heritage. Among the several conservation treatments available for carbonatic matrixes, the treatment with diammonium hydrogen phosphate (DAP, (NH4)2HPO4) is one of the most recent.1–6 DAP reacts with the carbonatic substrates, partially transforming the original minerals of the substrate (typically calcite) into newly formed phases (typically calcium phosphates).1,4,6–9When dealing with inorganic mineral treatments, the characterization of the newly formed phases and their location within the substrate is a key factor to determine the properties of the “stone-treatment” system. However, the investigation of the crystal chemistry and the spatial distribution of the newly formed phases is not straightforward, as they are composed of a complex mixture of micro- and submicro-crystals.4,5,10–16 Concerning the characterization of these phases, several analytical protocols have already been explored, including elemental analysis combined with electron microscopy (SEM-EDS), vibrational spectroscopies and neutron radiography or tomography.
SEM-EDS measurements investigate the marker elements of the reaction products (calcium and phosphorus and their characteristic Ca/P molar ratio), but the approach meets an intrinsic limitation of the “stone-treatment” system, namely the formation of non-stoichiometric partially substituted calcium phosphate phases, which alter the peculiar Ca/P molar ratio reported in the literature for specific crystalline phases.5,13
As for vibrational spectroscopies, both conventional FTIR (bulk analysis by scratching powder samples from different depths of treated samples6,17–19) and high resolution ATR μ-FTIR investigations (with a spatial resolution close to the diffraction limits13,20,21) have already been explored as analytical routes, but the identification of the phosphate compounds in complex mixtures is not straightforward even at the microscale.13
μ-Raman mapping has been extensively used to study the diffusion of inorganic treatments22,23 due to its high lateral resolution and inherent chemical selectivity. However, this technique suffers from strong limitations, e.g., fluorescence emitted by some compounds/lithotypes, weak Raman signals of the target mineral (such as phosphates) and extremely heterogeneous matrixes (such as stone substrates).5,11,14,24
Lastly, an imaging approach based on neutron radiography and/or tomography is able to provide indications of the distribution of the phases which contain high neutron cross-section elements, e.g. hydrogen,17,25–29 but it fails to identify the nature of the specific crystalline phases.
A diffractometric approach (X-ray diffraction, XRD) is expected to be the most suitable strategy to characterize the crystalline reaction products and their diffusion below the surface.30 Focusing on DAP treatments, recent studies carried out using X-ray powder diffraction (XRPD)4,11–13,17,31 investigated the phosphate phases formed on different carbonatic stone substrates by selecting sample powders at different depths from the DAP-treated surface. However, problems arise with conventional XRPD due to: (i) the prevalence of strong reflection peaks of calcite with respect to calcium phosphate phases, which are minor phases; (ii) the presence, in the bulk analysis, of a complex mixture of crystalline phases which have similar lattice parameters, as in the case of calcium phosphates, with the overlapping of their marker reflection peaks; (iii) the formation of non-stoichiometric, ionic substituted, calcium-deficient or poorly crystalline phases; (iv) the destructive nature of the investigation (scratching and grinding) coupled with the poor selectivity of the sampling procedure (conventional XRPD requires a relatively high amount of material, which involves a coarse sampling of treated stone).
The analytical outcome of XRD can be positively improved by using a synchrotron radiation (SR) source, as the perfectly monochromatic, collimated and highly brilliant beam provides lower detection limits and a higher spatial and spectral resolution with a micro-sized beamsize. In this view, a series of experiments have been carried out by SR-based grazing incidence XRD (GIXRD),10 with the investigation of phosphate phases formed on marble specimens and their interdependence with the minerals of the underlying marble portion. In the GIXRD experiments, the penetration depth of the X-rays within the stone matrix depends on several variables, including the elemental composition of the investigated materials, the X-ray energy and the incidence angle (Φ). On a given sample and by using a given X-ray energy, the principal variable which determines the penetration depth is Φ. This implies that when the measurements are carried out at very low Φ, the region investigated by the X-rays is confined to a superficial portion.
However, when dealing with consolidants, which reach a higher penetration depth than protective treatments and superficial coatings, a more complete overview on their diffusion is needed and a different XRD approach becomes necessary. SR micro-XRD in transmission geometry (SR-μTXRD) allows investigating the sample in transmission mode (proper samples, e.g., small fragments or thin sections, permit X-ray penetration into the bulk). In this way, the SR-μTXRD approach provides crystallochemical information of the crystalline phases by collecting diffraction patterns with a high spatial resolution of the whole investigated volume.32
SR-μTXRD is a well-established technique in materials science and soil chemistry,33 as well as in heritage science (e.g., in the investigation of pigments),32,34–38 to characterize polycrystalline materials even in complex phase associations without resorting to further micro-samplings or to destructive sample preparations.
However, no data are available on its utilization in conservation science to identify and localize the reaction products formed after inorganic-mineral conservation treatments on stone substrates.
Thus, here we report, for the first time, the application of SR-μTXRD for an in situ, spatially resolved and non-destructive study of the calcium phosphates formed as reaction products after a DAP-based consolidating treatment, aimed at exploring their possible phase changes within the sample and penetration depth. The goal is to explore the potential and limits of the SR-μTXRD method to provide unambiguous information on the nature, localization with respect to the substrate and possible orientations of crystalline reaction products formed by the conservation treatments. The analyses were performed on thin sections of porous carbonatic lithotype (the Noto yellowish limestone) specimens treated by poultice with 0.76 M DAP solutions, and the investigations were carried out considering the strong heterogeneity of the stone material and by exploring the formation of structurally similar crystalline phases. The feasibility of this innovative SR-μTXRD approach is demonstrated here in a quite challenging case-study.
2 Materials and methods
2.1 Materials
This study was carried out on prismatic specimens (5 × 5 × 2 cm3) of freshly quarried Noto limestone, a calcarenite outcropping in the Val di Noto (South-eastern Sicily) widely used as building and ornamental stones for the Baroque monuments.39–43 The Noto yellowish limestone variety was selected for the study among several varieties of limestones outcropping in the Iblean Plateau. The lithotype is a biomicrite42 mainly made of calcite, with secondary clay minerals, quartz and iron oxides, and high porosity (25–37%) in the range of micro-pores.40,43 The Noto limestone was consolidated by applying a poultice with a 0.76 M DAP water solution (DAP CAS number 7783-28-0, assay ≥99.0%, reagent grade; dry cellulose pulp MH 300 Phase, Italy; poultice ratio DAP solution: cellulose pulp 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) on the 5 × 5 cm2 external surface of the specimens. The treatment protocol (DAP molarity, application method) was defined, among the wide range of possibilities explored in the field,7,8 on the basis of our previous experiments,5,10,11,13,44 the literature18 and the restoration practices.45 The treatment timeframe is also a key point in DAP consolidation, as it favours the crystallization of specific crystalline phases and/or inhibits the formation of others;5 however, no data are available on the influence of the treatment duration on the DAP reaction with bulk Noto limestone. Then, two treatment times were selected (i.e., 2 h and 24 h), in order to study their influence on the composition and spatial distribution of the reaction products. During the treatment, the specimens (three for each treatment time) were wrapped in a plastic film to prevent the evaporation of the DAP solution. At the end of the treatment, the plastic film was removed and the specimens were dried at room temperature for 24 h with the poultice on top. Then, the poultice was removed and all the specimens were rinsed three times by immersion in deionised water and dried again.
:1) on the 5 × 5 cm2 external surface of the specimens. The treatment protocol (DAP molarity, application method) was defined, among the wide range of possibilities explored in the field,7,8 on the basis of our previous experiments,5,10,11,13,44 the literature18 and the restoration practices.45 The treatment timeframe is also a key point in DAP consolidation, as it favours the crystallization of specific crystalline phases and/or inhibits the formation of others;5 however, no data are available on the influence of the treatment duration on the DAP reaction with bulk Noto limestone. Then, two treatment times were selected (i.e., 2 h and 24 h), in order to study their influence on the composition and spatial distribution of the reaction products. During the treatment, the specimens (three for each treatment time) were wrapped in a plastic film to prevent the evaporation of the DAP solution. At the end of the treatment, the plastic film was removed and the specimens were dried at room temperature for 24 h with the poultice on top. Then, the poultice was removed and all the specimens were rinsed three times by immersion in deionised water and dried again.
Finally, the 5 × 5 × 2 cm3 specimens were cut into 3 × 3 × 2 cm3 specimens to avoid any possible “boundary effects” (dropping of the DAP solution or drying of the treatment) but preserving the treated surface. The 3 × 3 × 2 cm3 specimens were further cut into smaller samples (transversal cut: from the treated surface to the opposite side of the specimens, parallel to the diffusion direction of the treatment). Some of them, hereafter defined as cross sections, were used without any additional sample preparation (samples for the conventional X-ray powder diffraction); others were embedded in epoxy resin and prepared as polished cross sections (samples for SEM-EDS) and petrographic “thin” sections (thickness of ∼400–700 μm, samples for SR-μTXRD). All the sections were prepared by polishing the transversal side.
2.2 Methods
The elemental composition and the morphology of the specimens before and after the DAP consolidation were investigated using a JEOL 5910 LV scanning electron microscope (SEM) coupled with an IXRF-2000 energy dispersive X-ray spectrometer (EDS) on polished cross sections.The SR-μTXRD measurements were performed in two different experimental sessions in order to:
• Explore the penetration depth of the calcium phosphates phases in the whole porous stone matrix. The experiments were carried out at the XRD1 beamline46 of the Elettra – Synchrotron Trieste S.c.P.A. (Basovizza – Italy). The diffraction data were collected with a monochromatic beam λ = 0.688800(1) Å, in the 2ϑ angular range 1.5–50° and with an angular stepsize of 0.01°. The beamsize was 40 × 40 μm2 and the linear stepsize was 30 μm, providing an oversampling of ∼10 μm between two adjacent measurements.
• Explore the possible phase evolution of different calcium phosphates in the portion of stone material closer to the surface. The experiments were carried out at the ID15b beamline47 of the European Synchrotron Radiation Facility (ESRF) (Grenoble – France). The diffraction data were collected with a monochromatic beam λ = 0.410500(3) Å, in the 2ϑ angular range of 1.5–45° and with an angular stepsize of 0.01°. The measurements were carried out by using a smaller beamsize (10 × 10 μm2) and a linear stepsize of 200 μm, which avoid oversampling between two adjacent measurements.
The depth of the SR-μTXRD measurements was checked by the high spatial and zenithal resolution setup of the beamlines. The Rietveld refinement on a silicon standard with the GSAS package was used to estimate the uncertainty of the experimental SR wavelengths.
Both the XRPD and the SR-μTXRD investigations were carried out on three replicates for each specimen.
3 Results and discussion
The reaction of DAP solutions with the calcite of Noto limestone produces a slight dissolution of the substrate followed by crystallization of new phases on the reaction profile of carbonate grains. These newly formed phases form a shell, namely a crystal network, which grows on the reaction profile of calcite grains and between grain boundaries.5,11 As shown by the SEM-EDS morphological investigations and elemental analysis (Fig. 1), the DAP consolidating treatment causes a noteworthy crystallization of Ca- and P-bearing compounds in the matrix down to several millimetres from the treated surface. The EDS investigations carried out on several areas of the shell show that calcium phosphates exhibit a different Ca/P molar ratio, suggesting the formation of different crystalline phases. By using EDS, it is not possible to unambiguously define the nature of these phases because the calcium phosphates formed by the DAP treatments are generally non-stoichiometric and their crystal chemistry strongly depends on the reaction conditions5,11–13 | ||
| Fig. 1 SEM images (BS) collected on a polished cross section of Noto limestone treated with the DAP solution and showing the nucleation of the shell of calcium phosphates in a pore within the matrix (a) and details at higher magnification (b). The SEM images are collected at a depth of 3 mm. | ||
In order to explore the composition of the newly formed phosphate phases and their penetration depth, a set of XRD investigations were carried out.
Fig. 2 shows a comparison among the X-ray powder diffraction patterns, collected with a conventional lab instrument, of the treated and untreated Noto limestone at different depths. The XRPD patterns mainly show the peaks of calcite (foremost peaks at 3.85(d012), 3.03(d104), 2.49(d110), 2.28(d113), and 2.09(d202) Å), sometimes associated with quartz (peak located at 3.34(d101) Å), and of a newly formed calcium phosphate phase. As demonstrated by recent studies,11,13 the peaks located at 3.42 Å (d002) and 1.71 Å (d004) and the broad peak between 2.89 and 2.53 Å can be ascribed to a poorly crystalline and partially substituted hydroxyapatite (HAP, ideally Ca10(PO4)6(OH)2). The presence of octacalcium phosphate (OCP, Ca8(HPO4)2(PO4)4·5H2O), a calcium phosphate phase often associated with HAP after DAP treatments,4,10,11,13,17 cannot be confirmed due to the high intensity of the background that likely hides its possible marker peaks (peaks at 18.56 Å (d010), 9.36 Å (d020), 9.03 Å (d110) and 5.49 Å (d−101)).
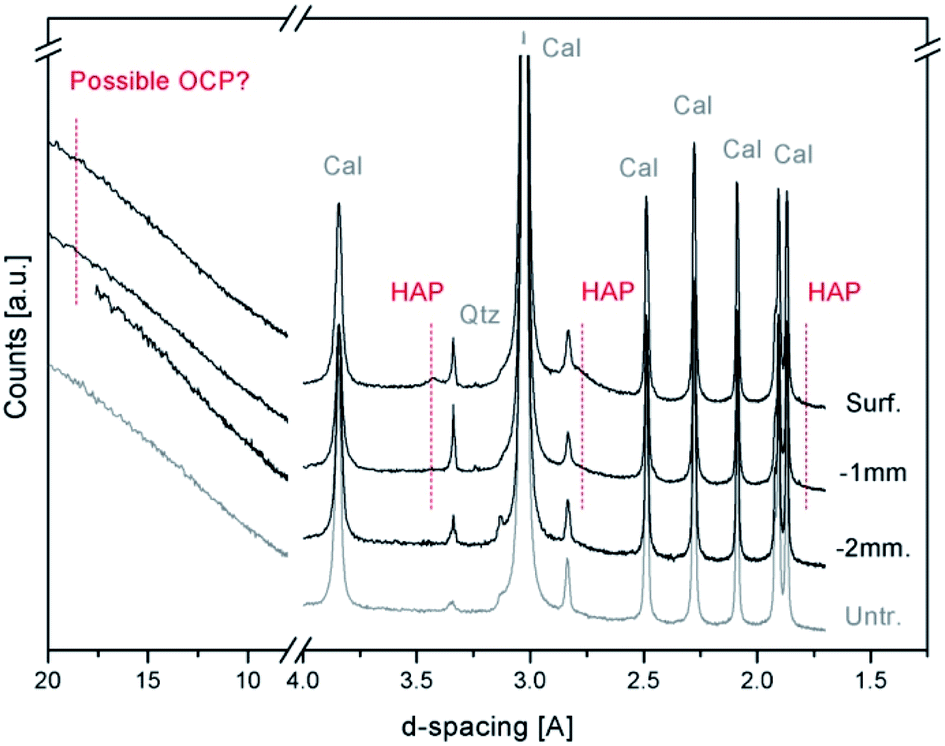 | ||
| Fig. 2 XRPD patterns of the untreated (Untr.) and DAP-treated Noto limestone specimens: powders were sampled on the surface (Surf.) and at 1 mm (−1 mm) and 2 mm (−2 mm) from the treated surface. The Bragg peaks of newly formed calcium phosphates (HAP = hydroxyapatite and possible OCP = octacalcium phosphate) are so weak that they are hardly detectable on the surface of the treated lithotype and in the most superficial portion of stone materials. Below a 2 mm depth, their detection becomes ambiguous. Cal = calcite, Qtz = quartz. | ||
In general, the newly formed phases are so poorly crystalline and in such a low fraction with respect to the calcite of the matrix that their XRD peaks are hardly detectable below a depth of 1–2 mm from the surface. As a consequence, the investigation with conventional XRPD is poorly exhaustive to identify the mineralogical composition or to determine the penetration depth.
The synchrotron radiation X-ray diffraction measurements carried out in transmission geometry on thin sections (SR-μTXRD) are used to overcome the analytical limits of the conventional XRPD approach. In particular, thanks to the micro-sized beam and the spatially resolved stepsize, the SR-μTXRD investigations allowed investigating, with a high resolution and selectivity, the nature of the reaction phases, their penetration depth and the possible phase variations along with the distance from the surface.
Fig. 3 shows the first sequence of SR-μTXRD measurements carried out with a beamsize of 40 × 40 μm2 and a stepsize of 30 μm, which provided an oversampling of ∼10 μm between two adjacent measurements. In this way, the SR-μTXRD measurements explore all the possible crystalline phases present along the penetration profile. For clarity's sake, the most representative XRD patterns are reported in Fig. 3.
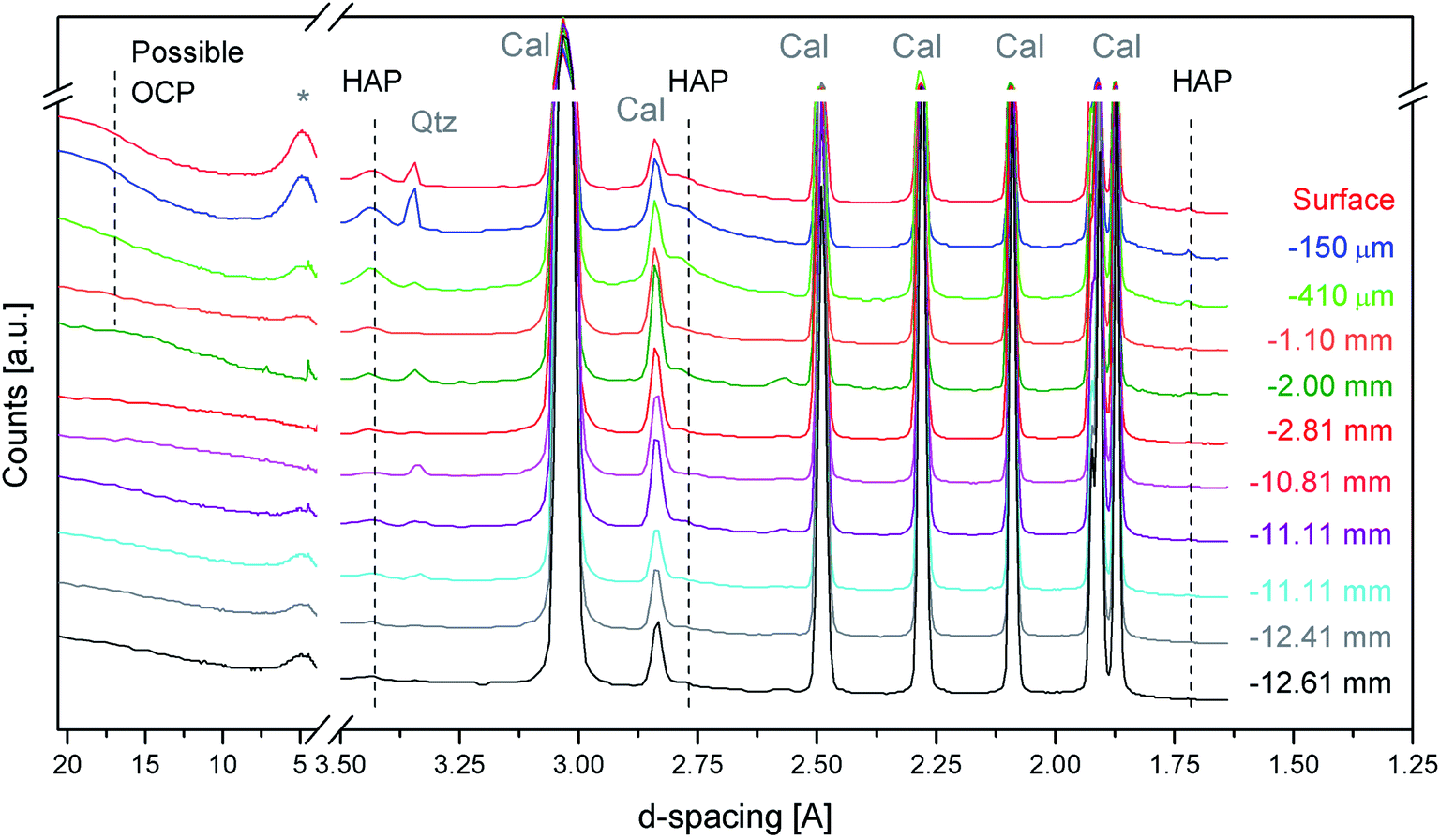 | ||
| Fig. 3 SR-μTXRD patterns of treated Noto limestone from the surface down to a 12.61 mm depth, showing the unambiguous presence HAP peaks. Possible peaks due to OCP are shown as well. Cal = calcite, Qtz = quartz, * = clay minerals. | ||
The SR-μTXRD patterns provide high quality data (in terms of spectral resolution, spatial resolution, and signal-to-noise ratio) and they allow detection of the Bragg peaks of newly formed calcium phosphates besides the minerals of the lithotype (calcite and other minor phases).
Focusing on secondary minerals of the stone matrix, the XRD patterns highlight the presence of kaolinite (main peak at 7.17–7.20 Å (d001)), or other clay minerals such as smectite/montmorillonite or illite (broad peaks at 4.48 Å and 2.57 Å), and quartz (4.25 Å (d100) and 3.34 Å). In all the cases, the intensity of their peaks is very low, and their relative abundance is randomised, most likely due to the compositional micro-heterogeneity of the stone matrix.
The characteristic diffraction peaks of HAP are visible at 3.43 Å (d002), 2.79 Å (d112) and 1.71 Å (d004) and they are unambiguously detected down to 10–11 mm from the treated surface. Below this depth, HAP peaks became less homogeneous and in many cases it is not easy to distinguish the peaks of possible HAP from the background fluctuations.
As regards the HAP peaks intensity, the SR-μTXRD patterns show a gradient: the signal is higher closer to the surface and progressively decreases as long as the data are collected from the inner portion of stone material.
This intensity gradient has to be considered only in terms of semi-quantitative evaluations and not quantitative, as the stone matrix is micro-heterogeneous and the HAP enrichment might have an inhomogeneous distribution (in pores and microcracks). However, this gradient can be correlated with the reactivity of the substrate and with the diffusion of the DAP solution. In fact, when the DAP poultice is applied to the stone specimens, the portion of stone material closer to the surface is reached by the highly reactive DAP solution and the crystallization of calcium phosphates is pronounced. As long as the reaction continues, the solution diffuses in the pore network by capillarity, reaching the inner portions, and it is progressively depleted of reagent. At the same time, a new fraction of the highly reactive solution is released from the poultice at the interface of the poultice/surface, penetrates through the portion of the stone material close to the surface (which experiences a new reaction with a fresh solution) and then is transmitted by capillarity toward the core and the bottom of the specimens.
Regarding the possible crystallization of other phosphate phases, in some SR-μTXRD patterns collected close to the surface there is a weak shoulder at ∼18.70 Å, which is compatible with OCP. However, the presence of a humped baseline in the initial spectral regions prevents the unambiguous identification of OCP.
The exhaustive characterization of the crystalline phases formed after DAP consolidation in the most external portion (i.e., the first hundreds of micrometers) of the treated stone materials is crucial, as this external region is the stone portion that will first interact with the environmental outdoor factors affected by, e.g., pollution and acid rains. It follows that a study of the diffraction patterns in the low theta region is needed in order to completely explore the possible presence of other phases. For this reason, a further set of investigations was carried out in the first millimetre of treated Noto limestone with a highly energetic SR beam, a smaller beamsize (10 × 10 μm2) and a stepsize of 200 μm. This allowed exploring the possible phase variations in this sample region without any overlapping between two adjacent measurements, thus avoiding any ambiguity in the spatial distribution of specific phases.
Fig. 4 shows the diffraction patterns collected at the ESRF for the treated Noto limestone, from the surface down to the first millimetre of the penetration depth. The high brilliance of the SR beam and the high sensitivity detector available at the beamline47 allowed collection of XRD patterns with a good signal-to-noise ratio and a very flat baseline and, therefore, identification of crystalline phases with high interplanar distances.
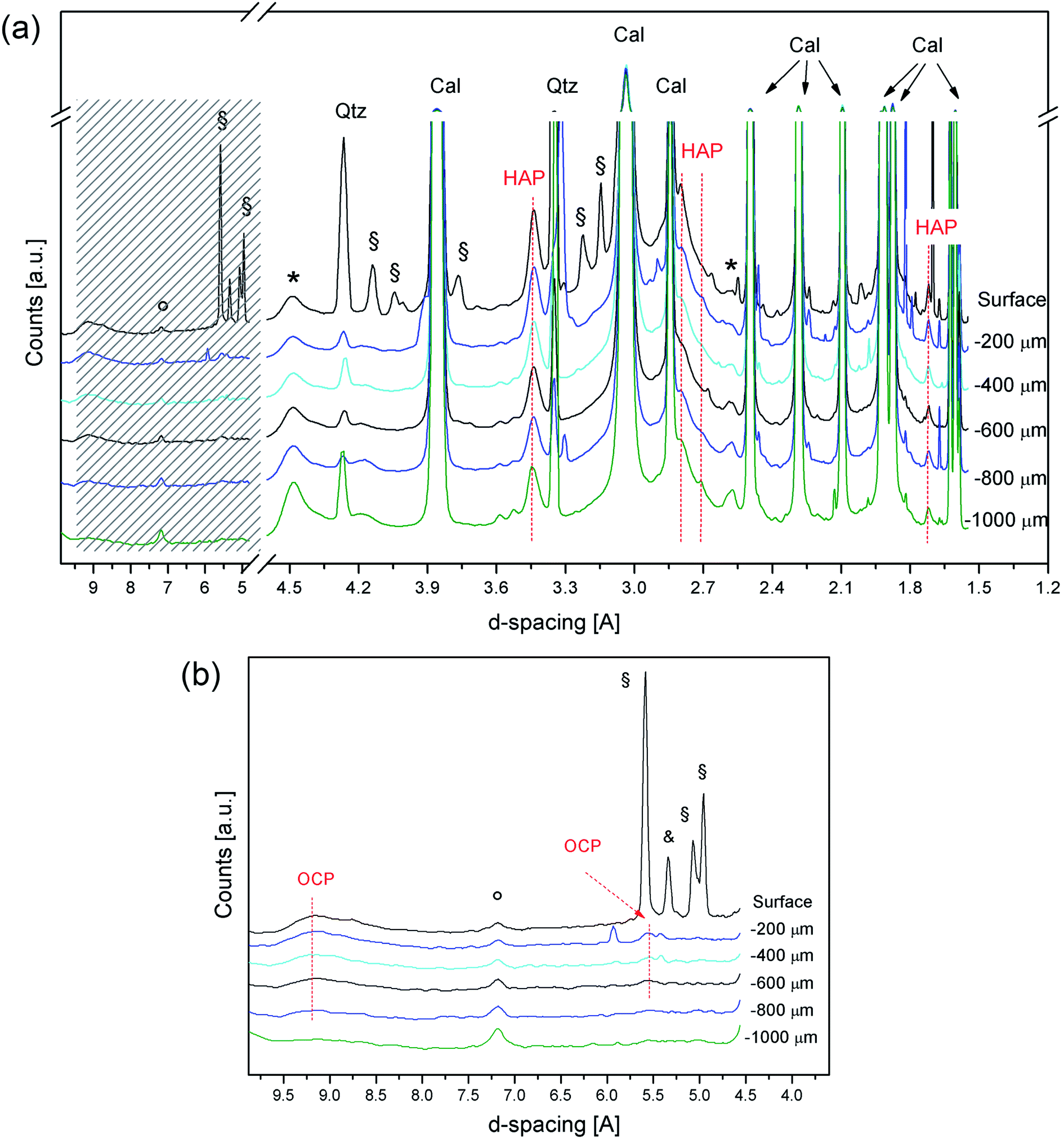 | ||
| Fig. 4 SR-μTXRD patterns at different depths from the surface of treated Noto limestone (a). Magnification of the SR-μTXRD patterns in the region at high interplanar distances (b). Cal = calcite, Qtz = quartz, HAP = hydroxyapatite, OCP = octacalcium phosphate, * = clay minerals (smectite/montmorillonite or illite), ° = kaolinite, § = unreacted DAP, & = unreacted ADP. HAP is present in all the patterns, while OCP is present from the surface down to 600–800 micrometers. | ||
The first difference from the previous measurements is the detection of peaks due to the reagent (DAP peaks at 5.59 Å, 5.06 Å, 5.01 Å shoulder, and 4.95 Å) and to ammonium dihydrogen phosphate (ADP, NH4H2PO4, peaks at 5.33 Å, 3.76 Å, and 2.37 Å), an acid by-product of the reaction. These two phases are detected only in the diffraction pattern referring to the surface, thus indicating that they are confined to the most superficial portion (i.e., a few micrometres).
DAP and ADP are highly soluble phases but they persist close to the surface of treated Noto limestone. This occurs on substrates with a high reactivity, such as micritic sedimentary matrixes,12 where the crystal network of Ca phosphates partially embeds ADP and DAP. In contrast, no soluble ammonium phosphates are detected after the DAP treatments on compact Carrara marble (unpublished results). Besides, the presence of ADP suggests a slight acidification of the solution in the most superficial portion of stone materials, a phenomenon previously hypothesised.13
Concerning calcium phosphates, HAP peaks are clearly detected in all the XRD patterns, thus confirming its widespread crystallization. Moreover, OCP is unequivocally detected as well from the peaks at 9.49–9.10 Å and at 5.51 Å in the patterns collected from 200 μm to 600–800 μm depths. The weak shoulder at 9.10 Å in the SR-μTXRD pattern collected in the most external portion suggests that OCP might also be present on the surface, even if the secondary peak at 5.51 Å is overlapped with a DAP peak. Below 600–800 μm depths, the identification of OCP becomes ambiguous.
In the course of crystal growth, OCP is probably formed as sub-micrometric crystals or contains structural defects and vacancies, as its XRD peaks are broad and weak. OCP is an acidic calcium phosphate and its formation is most likely due to the decrease of pH in the reaction environment during the last steps of the reaction.12,13 The lowering of the pH, also suggested by the detection of ADP, probably occurs in the whole matrix even though its effects are more evident in the outer part of the treated lithotypes, namely at the interface with the treatment poultice and at the interface with the air where the evaporation occurs. Furthermore, the formation of OCP (and HAP, as well) also depends on the Ca2+ availability and its fluctuations during the reaction. It follows that the formation of OCP, which has a lower Ca/P molar ratio than HAP,15 reflects a variation in the Ca2+ ionic strength in the DAP solution close to the treated surface, due to the “consumption” of calcium ions of the lithotype in this highly reactive stone region.
In any case, HAP and OCP always generate powder-like XRD patterns, with peak intensities comparable to those reported for standard reference materials. These features show that their crystallization does not follow preferential orientations or textures but the crystallites are randomly distributed in three dimensions within the stone matrix instead. In contrast, preferential orientations of crystallites should generate anomalous intensities of some given diffraction peaks.
The results, in terms of composition and localization of the crystalline phases, are similar between the specimens treated for 2 h or for 24 h, indicating that the crystallization and localization of OCP and HAP are irrespective of the treatment duration. This suggests that substantial results for stone conservation can be achieved even with a treatment duration shorter than 24 h, as long as the DAP poultice is left to dry (for 24 h) on the treated surface. A significant finding of the study is that, by varying the treatment duration, no other calcium phosphates are formed during the treatment.
Concerning the properties of the newly formed phases, OCP is slightly more soluble than HAP (Ksp 25 °C mol l−1; OCP 2.8 × 10−97; HAP 1.6 × 10−117)15,16 and it is a metastable phase. However, its crystallization in a mixture with HAP is not a limitation for the conservation of the treated stones, as: (i) both the phases are definitely less soluble than the calcite of the substrate (Ksp 25 °C mol l−1 4.8 × 10−9);4 (ii) OCP is a HAP precursor and it tends to hydrolyze and to topotaxially transform into HAP in case of pH variations and in the presence of water,15 frequent outdoor conditions for treated stones. Therefore, these data supply important indications of the diffusion of the DAP treatments, of reactivity of the substrate and of the evolution of the “stone matrix-conservation product” system, and they represent a scientific background for supporting the conservation of stone materials in pilot worksites.
4 Conclusions
For the first time, a novel analytical approach based on SR-μTXRD has been used to study cultural heritage stone materials. The experimental findings of this study demonstrate that the SR-μTXRD protocol can be successfully used to investigate the mineralogical composition and penetration depth of inorganic mineral treatment in stone matrixes with a high spatial resolution.The approach is non-destructive, which is extremely useful when studying heterogeneous stone matrixes, as the specimens can be used for further investigations (e.g., computed tomography or morphological studies by optical microscopy and SEM).
Furthermore, the developed SR-μTXRD protocol provides information about the volume irradiated and transmitted. This implies that, in spite of the micrometric beamsize used, the technique can be considered a bulk analysis as long as the optical path used by the X-ray beam through the sample contains a representative number of crystal grains. In this way, it averages the possible micro-heterogeneity of the stone matrix and overcomes the limits of point-by-point analysis and surface techniques. In addition, by further reducing the beamsize and preparing thinner sections, it could be possible to explore the reaction of inorganic treatment with an even higher spatial resolution. A similar approach could improve the study of conservation products crystallized within stones with heterogeneous composition or, e.g., the reactivity of carbonatic aggregates to treatments applied to mortars and plasters.
Limitations of the SR-μTXRD protocol include the inability to achieve quantitative data and ambiguous analyses in the presence of grains bigger than the beamsize or not randomly arranged; in this case, the collected patterns are not fully comparable to “powder-like” ones. Moreover, being an XRD technique, it might be blind to the presence of newly formed amorphous compounds.
Focusing on the broader impact of the study, the SR-μTXRD protocol represents a new analytical route of high potential in materials sciences. In fact, the SR-μTXRD approach can be used to characterize the penetration depth of a crystalline phase “A” (or more phases) in a matrix “B”, permitting new diffusion studies for a wide range of materials, including other inorganic treatments applied to natural and artificial stone materials (e.g., frescos and wall paintings, mortars and plasters, ceramic materials and cements, and stuccoes), as well as to painted stratigraphies, metals, and glasses, and the diffusion of their decay products (e.g., soluble salts, crusts, deposits, corrosion products, and patinas).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Elettra – Synchrotron Trieste S.c.P.A. and the European Synchrotron Radiation Facility (ESRF) for the allocation of experimental beamtime through scientific proposals. GDG and MM acknowledge the support of the Italian Ministry of Education (MIUR) through the project “Dipartimenti di Eccellenza 2018–2022”.References
- M. Matteini, C. Colombo, G. Botticelli, M. Casati, C. Conti, R. Negrotti, M. Realini and E. Possenti, in Built Heritage 2013 Monitoring Conservation Management, 2013, pp. 1278–1286 Search PubMed.
- E. Sassoni and E. Franzoni, in Built Heritage 2013 Monitoring Conservation Management, 2013, pp. 1287–1295 Search PubMed.
- E. Sassoni, S. Naidu and G. W. Scherer, J. Cult. Herit., 2011, 12, 346–355 CrossRef.
- M. Matteini, S. Rescic, F. Fratini and G. Botticelli, Int. J. Architect. Herit., 2011, 5, 717–736 CrossRef.
- E. Possenti, C. Colombo, D. Bersani, M. Bertasa, A. Botteon, C. Conti, P. P. Lottici and M. Realini, Microchem. J., 2016, 127, 79–86 CrossRef CAS.
- E. Sassoni, G. Graziani and E. Franzoni, Constr. Build. Mater., 2016, 102, 918–930 CrossRef CAS.
- E. Sassoni, Materials, 2018, 11, 557 CrossRef PubMed.
- E. Sassoni, RILEM Tech. Lett., 2017, 2, 14–19 CrossRef.
- E. Sassoni, G. Graziani and E. Franzoni, Constr. Build. Mater., 2016, 102, 931–942 CrossRef CAS.
- E. Possenti, C. Colombo, C. Conti, L. Gigli, M. Merlini, J. R. Plaisier, M. Realini and G. D. Gatta, Appl. Phys. A, 2018, 124, 383 CrossRef.
- E. Possenti, Inorganic products used in the Conservation of Cultural Heritage: interaction with carbonatic substrates and newly-formed crystalline phases, Ph.D. thesis, University of Milan, 2019.
- E. Possenti, C. Conti, G. D. Gatta, M. Realini and C. Colombo, Coatings, 2019, 9, 169 CrossRef.
- E. Possenti, C. Colombo, C. Conti, L. Gigli, M. Merlini, J. R. Plaisier, M. Realini, D. Sali and G. D. Gatta, Constr. Build. Mater., 2019, 195, 557–563 CrossRef CAS.
- E. Sassoni, G. Graziani and E. Franzoni, Mater. Des., 2015, 88, 1145–1157 CrossRef CAS.
- L. Wang and G. H. Nancollas, Chem. Rev., 2008, 108, 4628–4669 CrossRef CAS.
- S. V. Dorozhkin, J. Mater. Sci., 2007, 42, 1061–1095 CrossRef CAS.
- G. Graziani, C. Colombo, C. Conti, E. Possenti, E. Perelli Cippo, M. Realini and E. Sassoni, Constr. Build. Mater., 2018, 187, 238–247 CrossRef CAS.
- E. Franzoni, E. Sassoni and G. Graziani, J. Cult. Herit., 2015, 16, 173–184 CrossRef.
- G. Graziani, E. Sassoni, G. W. Scherer and E. Franzoni, Constr. Build. Mater., 2017, 148, 571–578 CrossRef CAS.
- N. Calore, A. Botteon, C. Colombo, A. Comunian, E. Possenti, M. Realini, D. Sali and C. Conti, Vib. Spectrosc., 2018, 98, 105–110 CrossRef CAS.
- M. Bertasa, E. Possenti, A. Botteon, C. Conti, A. Sansonetti, R. Fontana, J. Striova and D. Sali, Analyst, 2017, 142, 4801–4811 RSC.
- C. Conti, C. Colombo, D. Dellasega, M. Matteini, M. Realini and G. Zerbi, J. Cult. Herit., 2011, 12, 372–379 CrossRef.
- C. Conti, C. Colombo, M. Matteini, M. Realini and G. Zerbi, J. Raman Spectrosc., 2010, 41, 1254–1260 CrossRef CAS.
- I. Osticioli, G. Botticelli, P. Matteini, S. Siano, R. Pini and M. Matteini, J. Raman Spectrosc., 2017, 48, 966–971 CrossRef CAS.
- C. Conti, C. Colombo, G. Festa, J. Hovind, E. P. Cippo, E. Possenti and M. Realini, J. Cult. Herit., 2016, 19, 463–466 CrossRef.
- M. Realini, C. Colombo, C. Conti, F. Grazzi, E. Perelli Cippo and J. Hovind, Anal. Bioanal. Chem., 2017, 409, 6133–6139 CrossRef CAS PubMed.
- F. Hameed, B. Schillinger, A. Rohatsch, M. Zawisky and H. Rauch, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 605, 150–153 CrossRef CAS.
- F. Hameed, M. Zawisky and A. Rohatsch, in 2015 9th International Symposium on Image and Signal Processing and Analysis (ISPA), IEEE, 2015, pp. 108–113 Search PubMed.
- M. I. Prudêncio, M. A. Stanojev Pereira, J. G. Marques, M. I. Dias, L. Esteves, C. I. Burbidge, M. J. Trindade and M. B. Albuquerque, J. Archaeol. Sci., 2012, 39, 964–969 CrossRef.
- F. Vanmeert, D. Mudronja, S. Fazinic, K. Janssens and D. Tibljas, X-Ray Spectrom., 2013, 42, 256–261 CrossRef CAS.
- E. Molina, L. Rueda-Quero, D. Benavente, A. Burgos-Cara, E. Ruiz-Agudo and G. Cultrone, Constr. Build. Mater., 2017, 143, 298–311 CrossRef CAS.
- E. Pouyet, B. Fayard, M. Salomé, Y. Taniguchi, F. Sette and M. Cotte, Heritage Sci., 2015, 3, 1–16 CrossRef.
- L. Denaix, F. van Oort, M. Pernes and A. G. Jongmans, Clays Clay Miner., 1999, 47, 637–646 CrossRef CAS.
- J. Romero-Pastor, A. Duran, A. B. Rodríguez-Navarro, R. Van Grieken and C. Cardell, Anal. Chem., 2011, 83, 8420–8428 CrossRef CAS.
- E. Welcomme, P. Walter, P. Bleuet, J.-L. Hodeau, E. Dooryhee, P. Martinetto and M. Menu, Appl. Phys. A, 2007, 89, 825–832 CrossRef CAS.
- E. Pouyet, M. Cotte, B. Fayard, M. Salomé, F. Meirer, A. Mehta, E. S. Uffelman, A. Hull, F. Vanmeert, J. Kieffer, M. Burghammer, K. Janssens, F. Sette and J. Mass, Appl. Phys. A, 2015, 121, 967–980 CrossRef CAS.
- M. Radepont, W. de Nolf, K. Janssens, G. Van der Snickt, Y. Coquinot, L. Klaassen and M. Cotte, J. Anal. At. Spectrom., 2011, 26, 959–968 RSC.
- K. Janssens, M. Alfeld, G. Van der Snickt, W. De Nolf, F. Vanmeert, M. Radepont, L. Monico, J. Dik, M. Cotte, G. Falkenberg, C. Miliani and B. G. Brunetti, Annu. Rev. Anal. Chem., 2013, 6, 399–425 CrossRef CAS.
- M. F. La Russa, G. Barone, C. M. Belfiore, P. Mazzoleni and A. Pezzino, Environ. Earth Sci., 2011, 62, 1263–1272 CrossRef.
- G. Alessandrini, A. Bocci, R. Bugini, D. Emmi, R. Peruzzi and M. Realini, in 7th International Congress on Deterioration and Conservation of Stone, 1993, pp. 11–20 Search PubMed.
- C. UrzÌ and M. Realini, Int. Biodeterior. Biodegrad., 1998, 42, 45–54 CrossRef.
- G. Barone, V. Crupi, F. Longo, D. Majolino, P. Mazzoleni, S. Raneri, J. Teixeira and V. Venuti, J. Instrum., 2014, 1–9 Search PubMed.
- G. Barbera, G. Barone, V. Crupi, F. Longo, G. Maisano, D. Majolino, P. Mazzoleni, S. Raneri, J. Teixeira and V. Venuti, Eur. J. Mineral., 2014, 26, 189–198 CrossRef CAS.
- E. Possenti, C. Colombo, C. Conti, N. Marinoni, M. Merlini, R. Negrotti, M. Realini and G. D. Gatta, Mater. Charact., 2019, 154, 315–324 CrossRef CAS.
- D. Pittaluga, F. Fratini, A. Nielsen and S. Rescic, in Scienza e Beni Culturali XXVIII, Arcadia Ricerche, 2012, pp. 303–312 Search PubMed.
- A. Lausi, M. Polentarutti, S. Onesti, J. R. Plaisier, E. Busetto, G. Bais, L. Barba, A. Cassetta, G. Campi, D. Lamba, A. Pifferi, S. C. Mande, D. D. Sarma, S. M. Sharma and G. Paolucci, Eur. Phys. J. Plus, 2015, 130, 43 CrossRef.
- M. Merlini and M. Hanfland, High Pressure Res., 2013, 33, 511–522 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |