
A high-throughput and high peak capacity narrow-bore parallel segmented flow column strategy for the liquid chromatography-tandem mass spectrometry analysis of organic contaminants in water†

Abstract
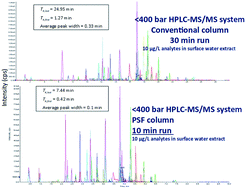
This study highlights the development of a high peak capacity, high-throughput (HTP) approach for a target list of 62 organic contaminants in an environmental river water matrix on a HPLC conventional 400 bar system. Key separation metrics were evaluated: (a) peak capacity, (b) total analysis time and (c) mobile phase consumption. An average peak width of 0.10 min with a total analysis time (inclusive of the column wash and re-equilibration) of 10 minutes revealed the increased productivity and performance of the parallel segmented flow (PSF) column technology HPLC-MS strategy, comparable to UHPLC peak widths and analysis times, achieved at a significantly lower backpressure (<400 bar). The operation of the PSF in a narrow-bore scale format (internal diameter of 2.1 mm) resulted in a conservative HPLC scale total mobile phase consumption of 15 mL for the column separation; only 5.5 mL of this volume was exposed to the ion source per injection. Mobile phase consumption was higher compared to that of UHPLC, but on the other hand it achieved higher peak capacity. Three representative compounds (atrazine, diclofenac and fluazuron) with differing retention and ionisation properties were studied in detail in terms of detection sensitivity. The majority of the ion ratios for the standards in the river water matrix were within ±30% of the average ion ratios. The largest ion suppression occurred for atrazine (−25% matrix effect), a pesticide notorious for poor ionization and poor peak shape issues. The lowest response transitions at 1 μg L−1 for the extracted ions in the river water matrix of atrazine, diclofenac and fluazuron had signal to noise ratios ≥3, with the exception of diclofenac where 5 μg L−1 was the lowest calibration level. The peak area's calibration curve slope and standard deviation in the detector's response determination of limit of quantification (LOQ) were between 5.2 and 30.4 μg L−1.
 Please wait while we load your content...
Please wait while we load your content...

